Bracon brevicornis Genome Showcases the Potential of Linked-Read Sequencing in Identifying a Putative Complementary Sex Determiner Gene

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Species Description and General Rearing
2.2. DNA Extraction
2.3. 10XGenomics Library Preparation and Sequencing
2.4. Assembly
2.5. Ab Initio Gene Finding and Protein Comparison
2.6. In Silico Identification of Feminizer as a Putative csd Locus
2.7. Microsynteny Analysis
2.8. Data Availability
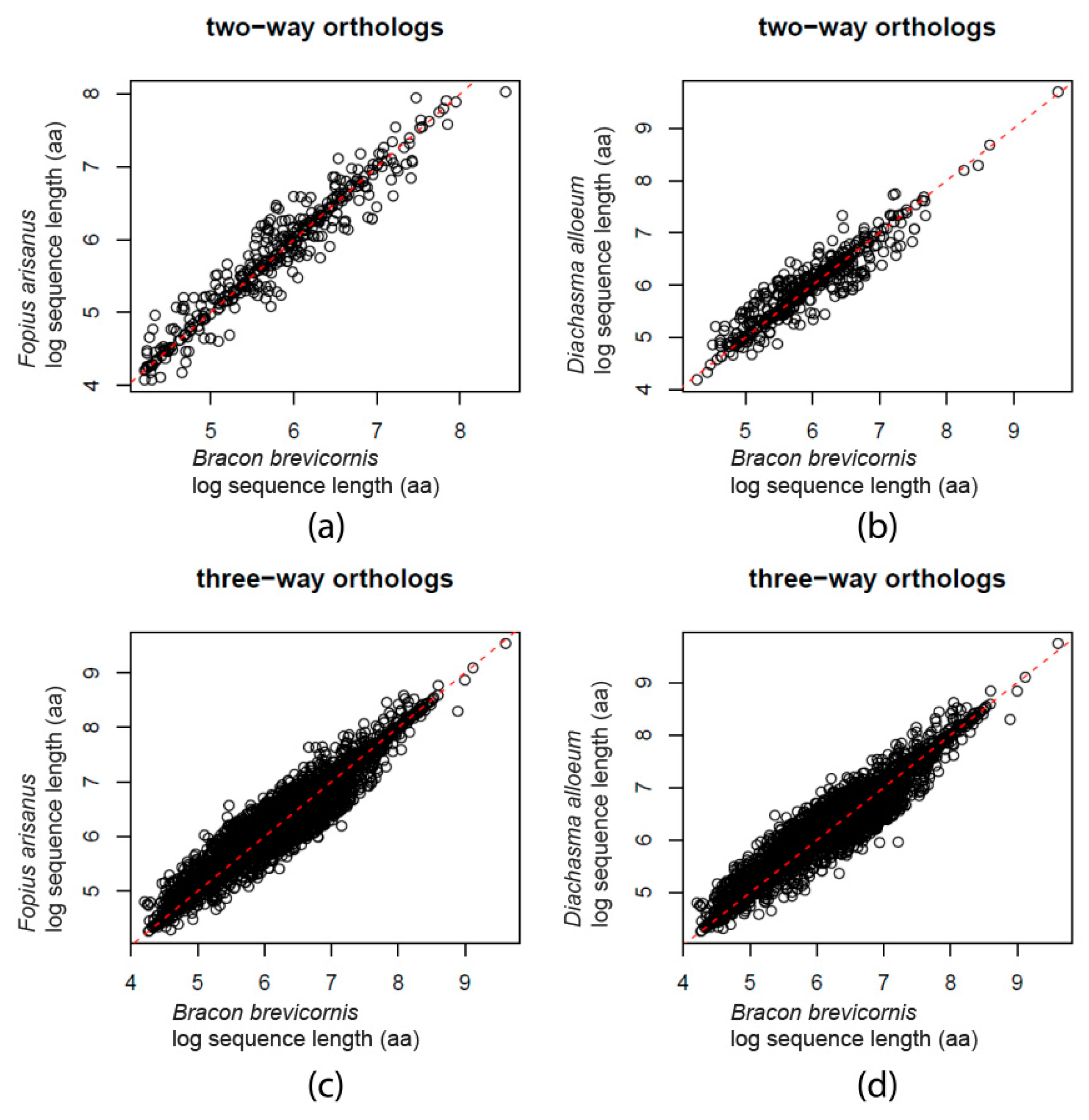
3. Results
3.1. Genome Assembly
3.2. Ab Initio Gene Finding and Protein Comparison
3.3. Identification of a Putative Feminizer Ortholog and Duplication Event
3.4. Synteny Analysis of Putative Fem Encoding Region
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Temerak, S.A. Longevity of Bracon brevicornis [Hym: Braconidae] adults as influenced by nourishment on artificial and natural foods. Entomophaga 1983, 28, 145–150. [Google Scholar] [CrossRef]
- Venkatesan, T.; Jalali, S.K.; Srinivasamurthy, K. Competitive interactions between Goniozus nephantidis and Bracon brevicornis, parasitoids of the coconut pest Opisina arenosella. Int. J. Pest. Manag. 2009, 55, 257–263. [Google Scholar] [CrossRef]
- Speicher, B.R.; Speicher, K.G. The Occurrence of Diploid Males in Habrobracon brevicornis. Am. Nat. 1940, 74, 379–382. [Google Scholar] [CrossRef]
- Narayanan, E.S.; Angalet, G.W.; Subba Rao, B.R.; D’Souza, G.I. Effect of refrigeration of the pupæ of microbracon brevicornis wesm. on the pigmentation of the adult. Nature 1954, 173, 503–504. [Google Scholar] [CrossRef]
- Puttarudriah, M.; Basavanna, G.P.C. A study on the identity of bracon hebetor say and bracon brevicornis wesmael (hymenoptera: Braconidae). Bull. Entomol. Res. 1956, 47, 183–191. [Google Scholar] [CrossRef]
- Kittel, R.N.; Maeto, K. Revalidation of Habrobracon brevicornis stat. rest. (Hymenoptera: Braconidae) Based on the CO1, 16S, and 28S Gene Fragments. J. Econ. Entomol. 2019, 112, 906–911. [Google Scholar] [CrossRef]
- Kares, E.A.; El-Sappagh, I.A.; Ebaid, G.H.; Sabra, I.M. Efficacy of releasing Bracon brevicornis wesm. (Hymenoptera: Braconidae) for controlling hibernated Ostrinia nubilalis (Hübner) and Sesamia cretica led. larvae in stored corn stalks. Egypt. J. Biol. Pest Control. 2010, 20, 155–159. [Google Scholar]
- Temerak, S.A. Host preferences of the parasitoid Bracon brevicornis Wesmael (Hym., Braconidae) and host sensitivity to its venom. Z. Angew. Entomol. 1983, 96, 37–41. [Google Scholar] [CrossRef]
- Srinivasan, T. Chandrikamohan Population growth potential of Bracon brevicornis Wesmael (Braconidae: Hymenoptera): A life table analysis. Acta Phytopathol. Entomol. Hung. 2017, 52, 123–130. [Google Scholar] [CrossRef]
- Villacañas de Castro, C.; Thiel, A. Resource-Dependent Clutch Size Decisions and Size-Fitness Relationships in a Gregarious Ectoparasitoid Wasp, Bracon brevicornis. J. Insect Behav. 2017, 30, 454–469. [Google Scholar] [CrossRef]
- Thiel, A.; Weeda, A.C. Haploid, diploid, and triploid—Discrimination ability against polyploid mating partner in the Parasitic Wasp, Bracon brevicornis (Hymenoptera: Braconidae). J. Insect Sci. 2014, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.; Weeda, A.C.; de Boer, J.G.; Hoffmeister, T.S. Genetic incompatibility drives mate choice in a parasitic wasp. Front. Zool. 2013, 10, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; van Achterberg, C. Systematics, Phylogeny, and Evolution of Braconid Wasps: 30 Years of Progress. Annu. Rev. Entomol. 2019, 64, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.M.; Crozier, R.H. Sex determination and population biology in the hymenoptera. Trends Ecol. Evol. 1995, 10, 281–286. [Google Scholar] [CrossRef]
- Heimpel, G.E.; de Boer, J.G. Sex Determination in the Hymenoptera. Annu. Rev. Entomol. 2008, 53, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.M. Sex determination in the Hymenoptera: A review of models and evidence. Heredity 1993, 71, 421–435. [Google Scholar] [CrossRef] [Green Version]
- Whiting, P.W.; Whiting, A.E. Diploid males from fertilized eggs in hymenoptera. Science (80-) 1925, 62, 437. [Google Scholar] [CrossRef]
- Clark, A.M.; Bertrand, H.A.; Smith, R.E. Life Span Differences between Haploid and Diploid Males of Habrobracon serinopae after Exposure as Adults to X Rays. Am. Nat. 1963, 97, 203–208. [Google Scholar] [CrossRef]
- Van Wilgenburg, E.; Driessen, G.; Beukeboom, L.W. Single locus complementary sex determination in Hymenoptera: An “unintelligent” design? Front. Zool. 2006, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Holloway, A.K.; Heimpel, G.E.; Strand, M.R.; Antolin, M.F. Survival of diploid males in Bracon sp. near hebetor (Hymenoptera: Braconidae). Ann. Entomol. Soc. Am. 1999, 92, 110–116. [Google Scholar] [CrossRef]
- Thiel, A. (University of Bremen, FB02, Institute of Ecology, Population and Evolutionary Ecology Group, Bremen, Germany). Personal communication, 2019. [Google Scholar]
- Whiting, P.W. Multiple alleles in sex determination of Habrobracon. J. Morphol. 1940, 66, 323–355. [Google Scholar] [CrossRef]
- Antolin, M.F.; Ode, P.J.; Heimpel, G.E.; O’Hara, R.B.; Strand, M.R. Population structure, mating system, and sex-determining allele diversity of the parasitoid wasp Habrobracon hebetor. Heredity 2003, 91, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Beye, M.; Hasselmann, M.; Fondrk, M.K.; Page, R.E.; Omholt, S.W. The gene csd is the primary signal for sexual development in the honeybee and encodes an SR-type protein. Cell 2003, 114, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Hasselmann, M.; Beye, M. Signatures of selection among sex-determining alleles of the honey bee. Proc. Natl. Acad. Sci. USA 2004, 101, 4888–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselmann, M.; Gempe, T.; Schiøtt, M.; Nunes-Silva, C.G.; Otte, M.; Beye, M. Evidence for the evolutionary nascence of a novel sex determination pathway in honeybees. Nature 2008, 454, 519–522. [Google Scholar] [CrossRef]
- Geuverink, E.; Beukeboom, L.W. Phylogenetic Distribution and Evolutionary Dynamics of the Sex Determination Genes doublesex and transformer in Insects. Sex. Dev. 2014, 8, 38–49. [Google Scholar] [CrossRef]
- Gempe, T.; Hasselmann, M.; Schiøtt, M.; Hause, G.; Otte, M.; Beye, M. Sex determination in honeybees: Two separate mechanisms induce and maintain the female pathway. PLoS Biol. 2009, 7, e1000222. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, S.; Colinet, D.; Poirié, M. Tracing back the nascence of a new sex-determination pathway to the ancestor of bees and ants. Nat. Commun. 2012, 3, 895. [Google Scholar] [CrossRef] [Green Version]
- Privman, E.; Wurm, Y.; Keller, L. Duplication and concerted evolution in a master sex determiner under balancing selection. Proc. R. Soc. B Biol. Sci. 2013, 280, 20122968. [Google Scholar] [CrossRef] [Green Version]
- Koch, V.; Nissen, I.; Schmitt, B.D.; Beye, M. Independent evolutionary origin of femparalogous genes and complementary sex determination in hymenopteran insects. PLoS ONE 2014, 9, e91883. [Google Scholar] [CrossRef]
- Biewer, M.; Schlesinger, F.; Hasselmann, M. The evolutionary dynamics of major regulators for sexual development among Hymenoptera species. Front. Genet. 2015, 6, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisenfeld, N.I.; Kumar, V.; Shah, P.; Church, D.M.; Jaffe, D.B. Direct determination of diploid genome sequences. Genome Res. 2017, 27, 757–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wührer, B.; (AMW Nützlinge GmbH, Pfungstadt, Germany). Personal communication, 2019.
- Chang, S.; Puryear, J.; Cairney, J. A simple and efficient method for isolating RNA from pine trees. Plant Mol. Biol. Rep. 1993, 11, 113–116. [Google Scholar] [CrossRef]
- Hosmani, P.S.; Flores-Gonzalez, M.; van de Geest, H.; Maumus, F.; Bakker, L.V.; Schijlen, E.; van Haarst, J.; Cordewener, J.; Sanchez-Perez, G.; Peters, S.; et al. An improved de novo assembly and annotation of the tomato reference genome using single-molecule sequencing, Hi-C proximity ligation and optical maps. bioRxiv 2019, 767764. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of genome assemblies [version 1; peer review: 2 approved with reservations]. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Acland, A.; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bryant, S.H.; Canese, K.; Church, D.M.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2014, 42, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Schatz, M.C.; Gurtowski, J.; Underwood, C.J.; Vurture, G.W.; Fang, H. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Li, X.Y.; van Achterberg, C.; Tan, J.C. Revision of the subfamily Opiinae (Hymenoptera, Braconidae) from Hunan (China), including thirty-six new species and two new genera. Zookeys 2013, 268, 1–186. [Google Scholar] [CrossRef] [Green Version]
- Geib, S.M.; Liang, G.H.; Murphy, T.D.; Sim, S.B. Whole Genome Sequencing of the Braconid Parasitoid Wasp Fopius arisanus, an Important Biocontrol Agent of Pest Tepritid Fruit Flies. G3 Genes Genomes Genet. 2017, 7, 2407–2411. [Google Scholar] [CrossRef] [Green Version]
- Tvedte, E.S.; Walden, K.K.O.; McElroy, K.E.; Werren, J.H.; Forbes, A.A.; Hood, G.R.; Logsdon, J.M.; Feder, J.L.; Robertson, H.M. Genome of the parasitoid wasp diachasma alloeum, an emerging model for ecological speciation and transitions to asexual reproduction. Genome Biol. Evol. 2019, 11, 2767–2773. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Solovyev, V. Statistical Approaches in Eukaryotic Gene Prediction. In Handbook of Statistical Genetics; Balding, D., Cannings, C., Bishop, M., Eds.; Wiley-Interscience: Hoboken, NJ, USA, 2007; pp. 97–159. ISBN 9780470061619. [Google Scholar]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Veltri, D.; Wight, M.M.; Crouch, J.A. SimpleSynteny: A web-based tool for visualization of microsynteny across multiple species. Nucleic Acids Res. 2016, 44, W41–W45. [Google Scholar] [CrossRef]
- De Boer, J.G.; (Department of Terrestrial Ecology, Netherlands Institute of Ecology, Wageningen, The Netherlands). Personal communication, 2019.
- Wang, Z.; Liu, Z.; Wu, X.; Yan, W.; Zeng, Z. Polymorphism analysis of csd gene in six Apis mellifera subspecies. Mol. Biol. Rep. 2012, 39, 3067–3071. [Google Scholar] [CrossRef]
- Verhulst, E.C.; van de Zande, L.; Beukeboom, L.W. Insect sex determination: It all evolves around transformer. Curr. Opin. Genet. Dev. 2010, 20, 376–383. [Google Scholar] [CrossRef] [Green Version]
- Hediger, M.; Henggeler, C.; Meier, N.; Perez, R.; Saccone, G.; Bopp, D. Molecular Characterization of the Key Switch F Provides a Basis for Understanding the Rapid Divergence of the Sex-Determining Pathway in the Housefly. Genetics 2010, 184, 155–170. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.I.; Salazar, P.A.; Kučka, M.; Davies, R.W.; Dréau, A.; Aldás, I.; Power, O.B.; Nadeau, N.J.; Bridle, J.R.; Rolian, C.; et al. Haplotype tagging reveals parallel formation of hybrid races in two butterfly species. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dechaud, C.; Volff, J.N.; Schartl, M.; Naville, M. Sex and the TEs: Transposable elements in sexual development and function in animals. Mob. DNA 2019, 10, 42. [Google Scholar] [CrossRef]
- Geuverink, E.; Kraaijeveld, K.; van Leussen, M.; Chen, F.; Pijpe, J.; Linskens, M.H.K.; Beukeboom, L.W.; van de Zande, L. Evidence for involvement of a transformer paralogue in sex determination of the wasp Leptopilina clavipes. Insect Mol. Biol. 2018, 27, 780–795. [Google Scholar] [CrossRef]
- Jia, L.-Y.; Xiao, J.-H.; Xiong, T.-L.; Niu, L.-M.; Huang, D.-W. The transformer genes in the fig wasp C eratosolen solmsi provide new evidence for duplications independent of complementary sex determination. Insect Mol. Biol. 2016, 25, 191–201. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferguson, K.B.; Pannebakker, B.A.; Centurión, A.; van den Heuvel, J.; Nieuwenhuis, R.; Becker, F.F.M.; Schijlen, E.; Thiel, A.; Zwaan, B.J.; Verhulst, E.C. Bracon brevicornis Genome Showcases the Potential of Linked-Read Sequencing in Identifying a Putative Complementary Sex Determiner Gene. Genes 2020, 11, 1390. https://doi.org/10.3390/genes11121390
Ferguson KB, Pannebakker BA, Centurión A, van den Heuvel J, Nieuwenhuis R, Becker FFM, Schijlen E, Thiel A, Zwaan BJ, Verhulst EC. Bracon brevicornis Genome Showcases the Potential of Linked-Read Sequencing in Identifying a Putative Complementary Sex Determiner Gene. Genes. 2020; 11(12):1390. https://doi.org/10.3390/genes11121390
Chicago/Turabian StyleFerguson, Kim B., Bart A. Pannebakker, Alejandra Centurión, Joost van den Heuvel, Ronald Nieuwenhuis, Frank F. M. Becker, Elio Schijlen, Andra Thiel, Bas J. Zwaan, and Eveline C. Verhulst. 2020. "Bracon brevicornis Genome Showcases the Potential of Linked-Read Sequencing in Identifying a Putative Complementary Sex Determiner Gene" Genes 11, no. 12: 1390. https://doi.org/10.3390/genes11121390