Molecular Epidemiology and Evolution of European Bat Lyssavirus 2

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
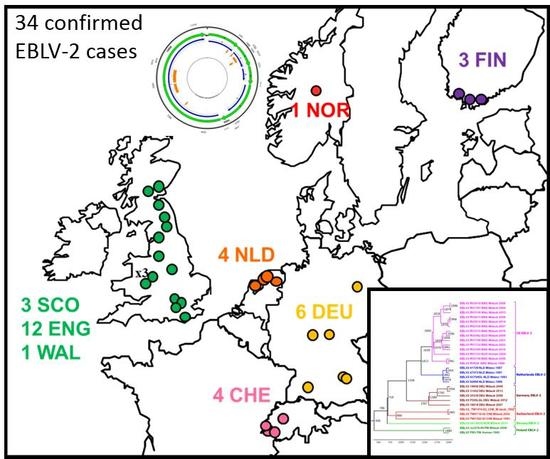
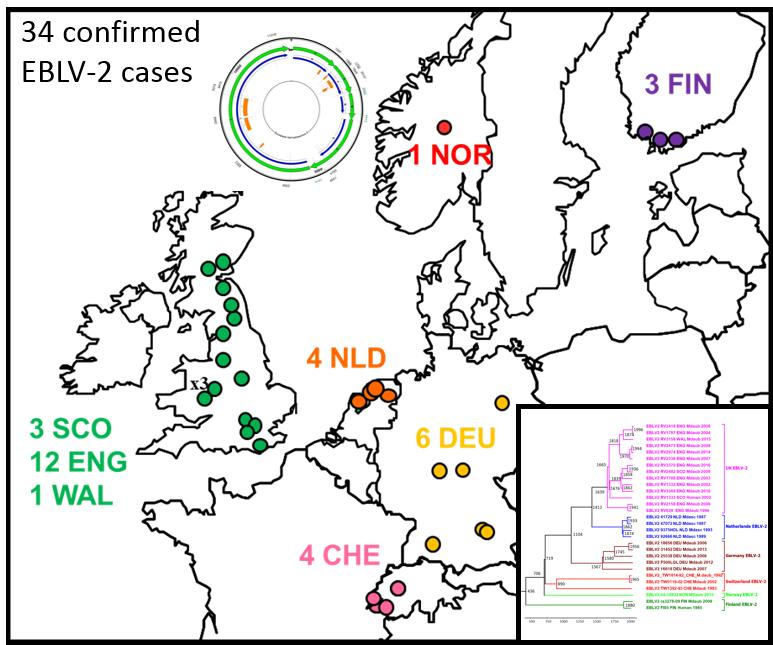
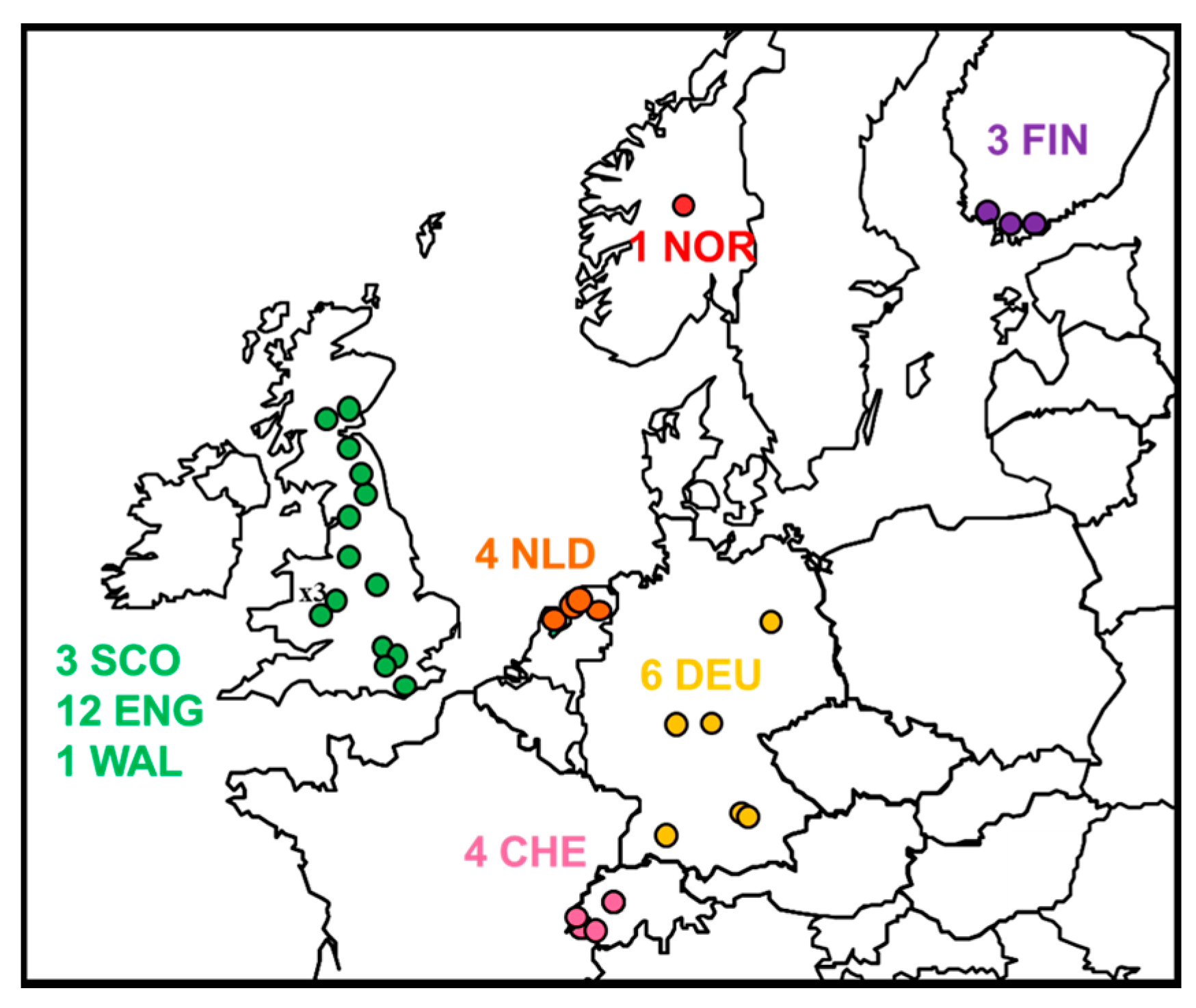
2.1. Review of Reported Cases
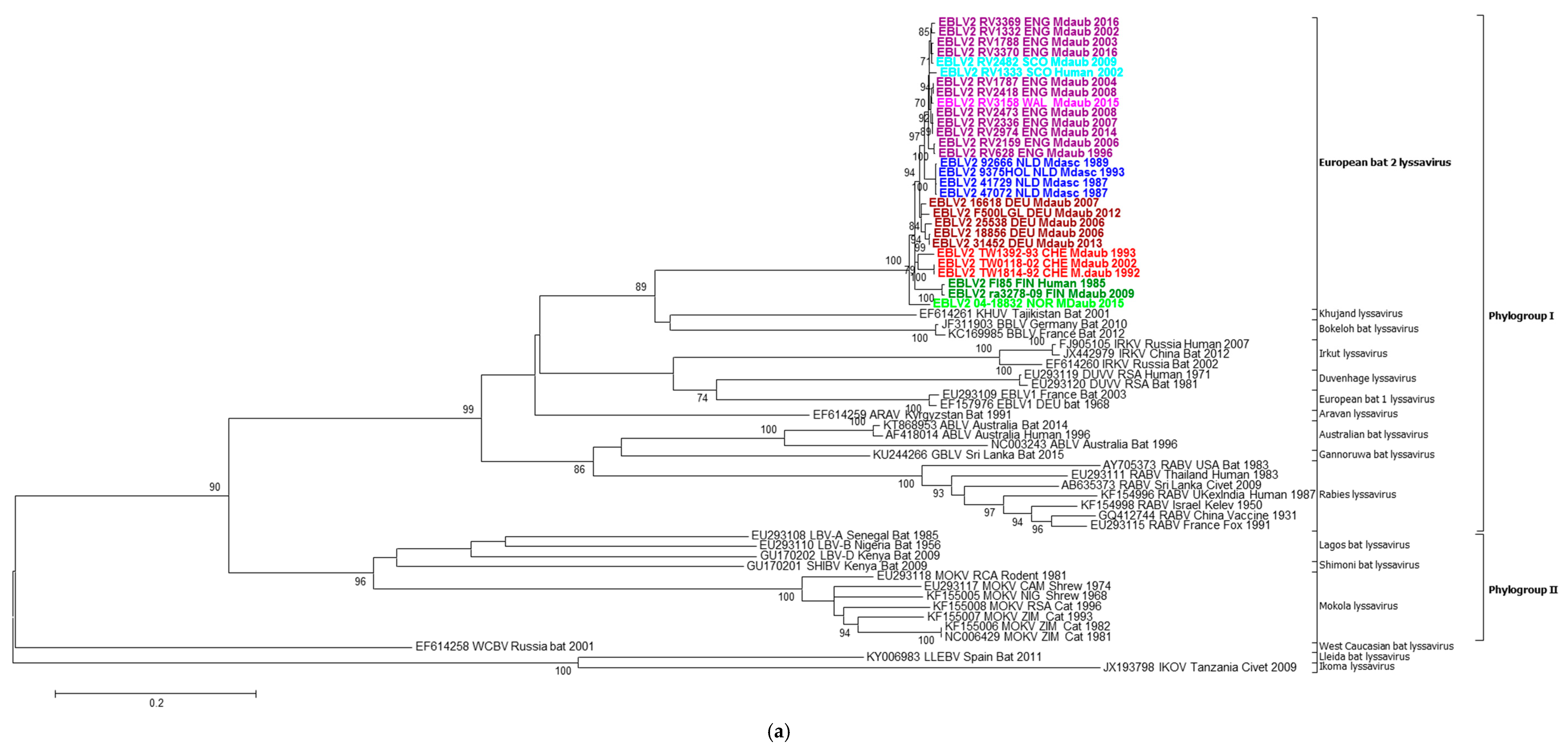
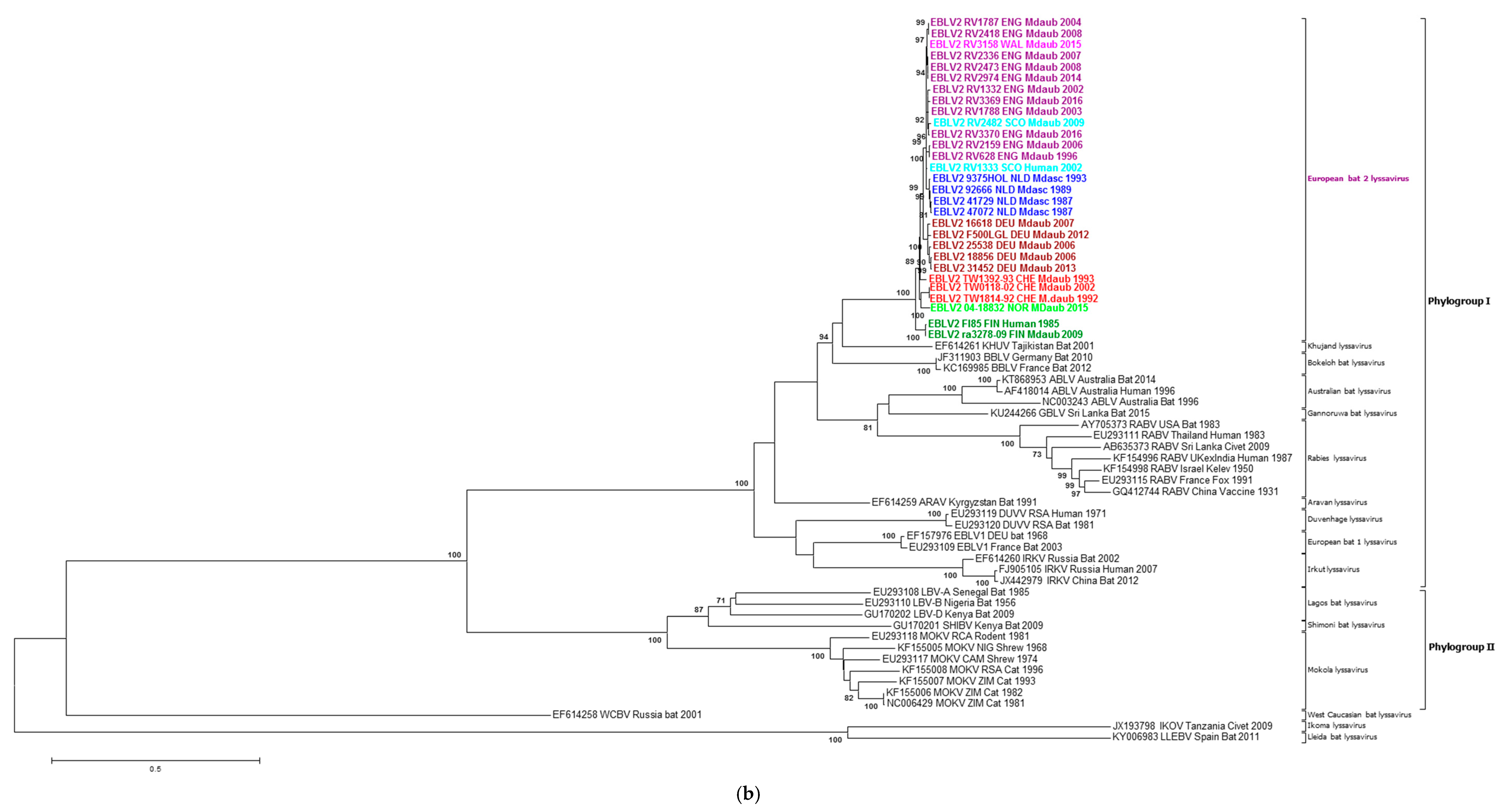
2.2. Phylogenetic Analysis
2.3. Intra-Roost Variation
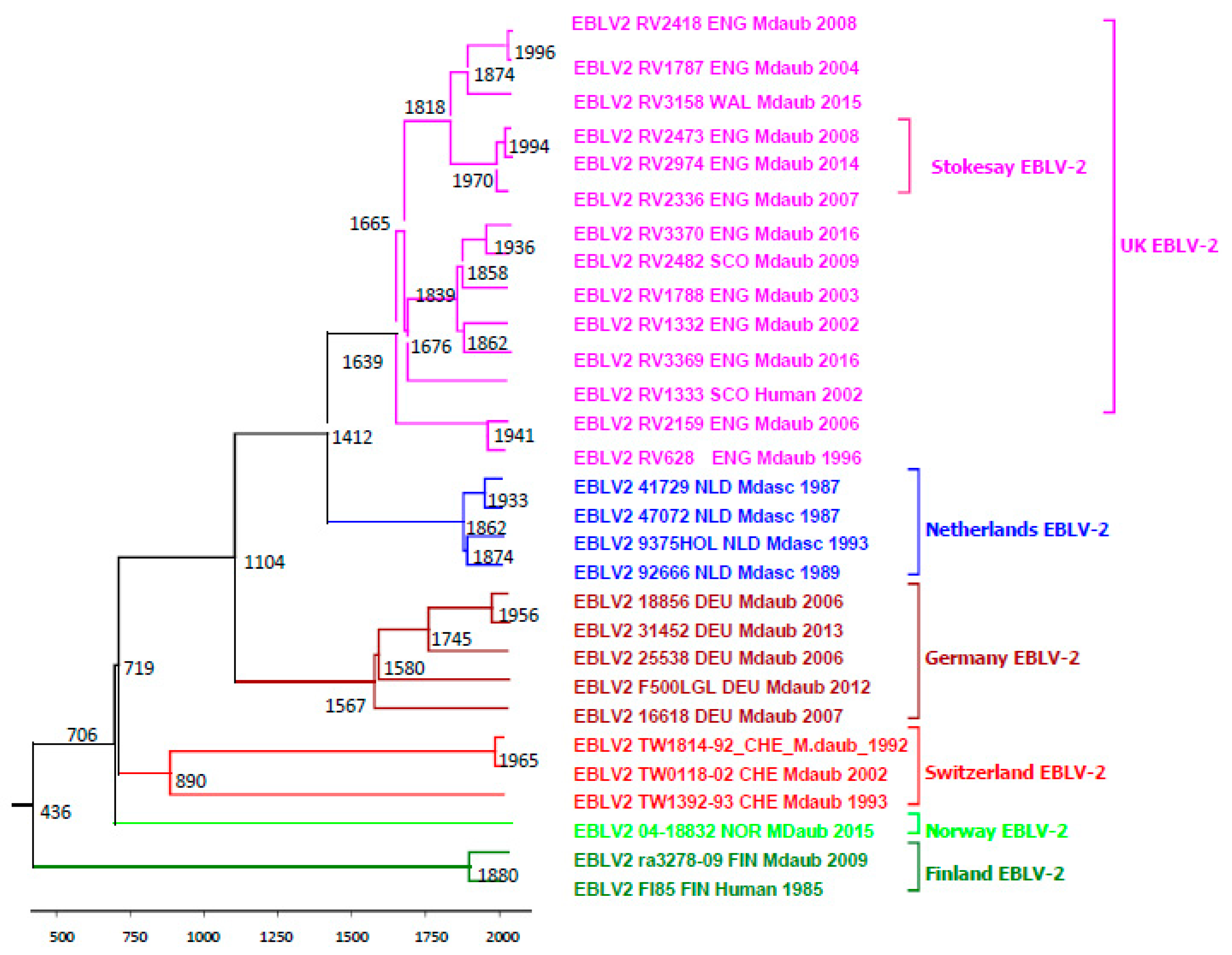
2.4. Evolutionary Analysis of EBLV-2
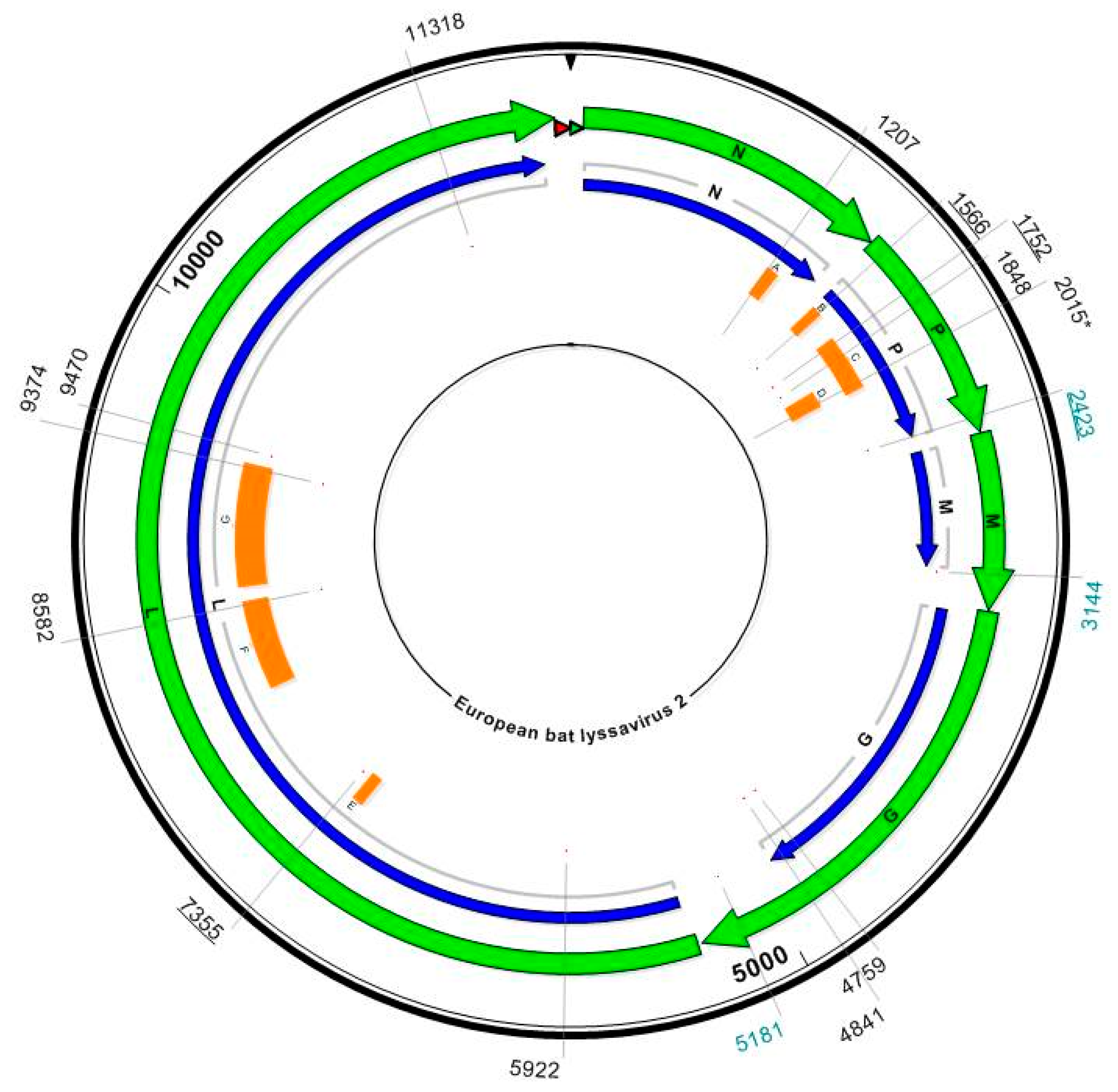
3. Materials and Methods
3.1. Next Generation Sequencing (NGS)
3.2. Mapping to Reference Sequence to Obtain Complete Genome Sequences
3.3. Evolutionary Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses (ICTV). Virus Taxonomy: The Classification and Nomenclature of Viruses. The Online (10th) Report of the ICTV. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 28 November 2017).
- Arechiga Ceballos, N.; Vazquez Moron, S.; Berciano, J.M.; Nicolas, O.; Aznar Lopez, C.; Juste, J.; Rodriguez Nevado, C.; Aguilar Setien, A.; Echevarria, J.E. Novel lyssavirus in bat, Spain. Emerg. Infect. Dis. 2013, 19, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardena, P.S.; Marston, D.A.; Ellis, R.J.; Wise, E.L.; Karawita, A.C.; Breed, A.C.; McElhinney, L.M.; Johnson, N.; Banyard, A.C.; Fooks, A.R. Lyssavirus in Indian flying foxes, Sri Lanka. Emerg. Infect. Dis. 2016, 22, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Banyard, A.C.; Hayman, D.; Johnson, N.; McElhinney, L.; Fooks, A.R. Bats and lyssaviruses. Adv. Virus Res. 2011, 79, 239–289. [Google Scholar] [PubMed]
- Botvinkin, A.D.; Poleschuk, E.M.; Kuzmin, I.V.; Borisova, T.I.; Gazaryan, S.V.; Yager, P.; Rupprecht, C.E. Novel lyssaviruses isolated from bats in Russia. Emerg. Infect. Dis. 2003, 9, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- Freuling, C.M.; Beer, M.; Conraths, F.J.; Finke, S.; Hoffmann, B.; Keller, B.; Kliemt, J.; Mettenleiter, T.C.; Muhlbach, E.; Teifke, J.P.; et al. Novel lyssavirus in Natterer’s bat, Germany. Emerg. Infect. Dis. 2011, 17, 1519–1522. [Google Scholar] [CrossRef] [PubMed]
- Picard-Meyer, E.; Servat, A.; Robardet, E.; Moinet, M.; Borel, C.; Cliquet, F. Isolation of Bokeloh bat lyssavirus in Myotis nattereri in France. Arch. Virol. 2013, 158, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Eggerbauer, E.; Troupin, C.; Passior, K.; Pfaff, F.; Hoper, D.; Neubauer-Juric, A.; Haberl, S.; Bouchier, C.; Mettenleiter, T.C.; Bourhy, H.; et al. The recently discovered bokeloh bat lyssavirus: Insights into its genetic heterogeneity and spatial distribution in Europe and the population genetics of its primary host. Adv. Virus Res. 2017, 99, 199–232. [Google Scholar] [PubMed]
- Mohr, W. Die Tollwut. Med. Klin. 1957, 52, 1057–1060. [Google Scholar] [PubMed]
- Lumio, J.; Hillbom, M.; Roine, R.; Ketonen, L.; Haltia, M.; Valle, M.; Neuvonen, E.; Lahdevirta, J. Human rabies of bat origin in Europe. Lancet 1986, 1. [Google Scholar] [CrossRef]
- Bourhy, H.; Kissi, B.; Lafon, M.; Sacramento, D.; Tordo, N. Antigenic and molecular characterization of bat rabies virus in Europe. J. Clin. Microbiol. 1992, 30, 2419–2426. [Google Scholar] [PubMed]
- Schatz, J.; Fooks, A.R.; McElhinney, L.; Horton, D.; Echevarria, J.; Vazquez-Moron, S.; Kooi, E.A.; Rasmussen, T.B.; Muller, T.; Freuling, C.M. Bat rabies surveillance in Europe. Zoonoses Public Health 2013, 60, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Brookes, S.M.; Aegerter, J.N.; Smith, G.C.; Healy, D.M.; Jolliffe, T.A.; Swift, S.M.; Mackie, I.J.; Pritchard, J.S.; Racey, P.A.; Moore, N.P.; et al. European bat lyssavirus in Scottish bats. Emerg. Infect. Dis. 2005, 11, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Amengual, B.; Bourhy, H.; Lopez-Roig, M.; Serra-Cobo, J. Temporal dynamics of European bat lyssavirus type 1 and survival of Myotis myotis bats in natural colonies. PLoS ONE 2007, 2, e566. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; Banyard, A.C.; McElhinney, L.M.; Freuling, C.M.; Finke, S.; de Lamballerie, X.; Müller, T.; Fooks, A.R. The lyssavirus host-specificity conundrum—Rabies virus—The exception not the rule. Curr. Opin. Virol. 2018, 28, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Badrane, H.; Tordo, N. Host switching in lyssavirus history from the chiroptera to the carnivora orders. J. Virol. 2001, 75, 8096–8104. [Google Scholar] [CrossRef] [PubMed]
- Vos, A.; Kaipf, I.; Denzinger, A.; Fooks, A.R.; Johnson, N.; Müller, T. European bat lyssaviruses—An ecological enigma. Acta Chiropterol. 2007, 9, 283–296. [Google Scholar] [CrossRef]
- Davis, P.L.; Holmes, E.C.; Larrous, F.; Van der Poel, W.H.; Tjornehoj, K.; Alonso, W.J.; Bourhy, H. Phylogeography, population dynamics, and molecular evolution of European bat lyssaviruses. J. Virol. 2005, 79, 10487–10497. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, L.M.; Marston, D.A.; Leech, S.; Freuling, C.M.; van der Poel, W.H.; Echevarria, J.; Vazquez-Moron, S.; Horton, D.L.; Muller, T.; Fooks, A.R. Molecular epidemiology of bat lyssaviruses in Europe. Zoonoses Public Health 2013, 60, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Jakava-Viljanen, M.; Nokireki, T.; Sironen, T.; Vapalahti, O.; Sihvonen, L.; HuoVilainen, A. Evolutionary trends of European bat lyssavirus type 2 including genetic characterization of Finnish strains of human and bat origin 24 years apart. Arch. Virol. 2015, 160, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Davies, P.; Lawrie, A. The rabies viruses of bats. Vet. Microbiol. 1990, 23, 165–174. [Google Scholar] [CrossRef]
- Kappeler, A. Bat rabies surveillance in Europe. Rabies Bull. Eur. 1989, 13, 12–13. [Google Scholar]
- Johnson, N.; Vos, A.; Neubert, L.; Freuling, C.; Mansfield, K.L.; Kaipf, I.; Denzinger, A.; Hicks, D.; Nunez, A.; Franka, R.; et al. Experimental study of European bat lyssavirus type-2 infection in Daubenton’s bats (Myotis daubentonii). J. Gen. Virol. 2008, 89, 2662–2672. [Google Scholar] [CrossRef] [PubMed]
- Rabies Bulletin Europe (RBE). Quarterly rabies cases reported in Europe. Rabies Bull. Eur. 1987, 11, 1–42. [Google Scholar]
- Megali, A.; Yannic, G.; Zahno, M.L.; Brugger, D.; Bertoni, G.; Christe, P.; Zanoni, R. Surveillance for European bat lyssavirus in Swiss bats. Arch. Virol. 2010, 155, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Whitby, J.E.; Heaton, P.R.; Black, E.M.; Wooldridge, M.; McElhinney, L.M.; Johnstone, P. First isolation of a rabies-related virus from a Daubenton’s bat in the United Kingdom. Vet. Rec. 2000, 147, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Selden, D.; Parsons, G.; Healy, D.; Brookes, S.M.; McElhinney, L.M.; Hutson, A.M.; Fooks, A.R. Isolation of a European bat lyssavirus type 2 from a Daubenton’s bat in the United Kingdom. Vet. Rec. 2003, 152, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; McElhinney, L.M.; Pounder, D.J.; Finnegan, C.J.; Mansfield, K.; Johnson, N.; Brookes, S.M.; Parsons, G.; White, K.; McIntyre, P.G.; et al. Case report: Isolation of a European bat lyssavirus type 2a from a fatal human case of rabies encephalitis. J. Med. Virol. 2003, 71, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; McElhinney, L.M.; Marston, D.A.; Selden, D.; Jolliffe, T.A.; Wakeley, P.R.; Johnson, N.; Brookes, S.M. Identification of a European bat lyssavirus type 2 in a Daubenton’s bat found in Staines, Surrey, UK. Vet. Rec. 2004, 155, 434–435. [Google Scholar] [PubMed]
- Fooks, A.R.; Selden, D.; Brookes, S.M.; Johnson, N.; Marston, D.A.; Jolliffe, T.A.; Wakeley, P.R.; McElhinney, L.M. Identification of a European bat lyssavirus type 2 in a Daubenton’s bat found in Lancashire. Vet. Rec. 2004, 155, 606–607. [Google Scholar] [PubMed]
- Fooks, A.R.; Marston, D.; Parsons, G.; Earl, D.; Dicker, A.; Brookes, S.M. Isolation of EBLV-2 in a Daubenton’s bat (Myotis daubentonii) found in Oxfordshire. Vet. Rec. 2006, 159, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Freuling, C.M.; Kliemt, J.; Schares, S.; Heidecke, D.; Driechciarz, R.; Schatz, J.; Muller, T. Detection of European bat lyssavirus 2 (EBLV-2) in a Daubenton’s bat (Myotis daubentonii) from Magdeburg, Germany. Berl. Munchener Tierarztliche Wochenschr. 2012, 125, 255–258. [Google Scholar]
- Schatz, J.; Ohlendorf, B.; Busse, P.; Pelz, G.; Dolch, D.; Teubner, J.; Encarnacao, J.A.; Muhle, R.U.; Fischer, M.; Hoffmann, B.; et al. Twenty years of active bat rabies surveillance in Germany: A detailed analysis and future perspectives. Epidemiol. Infect. 2014, 142, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Freuling, C.; Grossmann, E.; Conraths, F.J.; Schameitat, A.; Kliemt, J.; Auer, E.; Greiser-Wilke, I.; Muller, T. First isolation of EBLV-2 in Germany. Vet. Microbiol. 2008, 131, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Mansfield, K.; Marston, D.A.; Johnson, N.; Pajamo, K.; O’Brien, N.; Black, C.; McElhinney, L.M.; Fooks, A.R. Isolation of european bat lyssavirus type 2 from a Daubenton’s bat (Myotis daubentonii) in Shropshire. Vet. Rec. 2007, 161, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Pajermo, K.; Harkess, G.; Goddard, T.; Marston, D.; McElhinney, L.; Johnson, N.; Fooks, A.R. Isolation of european bat lyssavirus type 2 (EBLV-2) in a Daubenton’s bat in the UK with a minimum in Cubation period of 9 months. Rabies Bull. Eur. 2008, 32, 6–8. [Google Scholar]
- Banyard, A.C.; Johnson, N.; Voller, K.; Hicks, D.; Nunez, A.; Hartley, M.; Fooks, A.R. Repeated detection of european bat lyssavirus type 2 in dead bats found at a single roost site in the UK. Arch. Virol. 2009, 154, 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Horton, D.L.; Voller, K.; Haxton, B.; Johnson, N.; Leech, S.; Goddard, T.; Wilson, C.; McElhinney, L.M.; Fooks, A.R. European bat lyssavirus type 2 in a Daubenton’s bat in Scotland. Vet. Rec. 2009, 165, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Jakava-Viljanen, M.; Lilley, T.; Kyheroinen, E.M.; HuoVilainen, A. First encounter of European bat lyssavirus type 2 (EBLV-2) in a bat in Finland. Epidemiol. Infect. 2010, 138, 1581–1585. [Google Scholar] [CrossRef] [PubMed]
- Neubauer-Juric, A.; Krebs, S.; Schille, A.-U.; Schürmann, E.-M.; Blum, H. Tollwut in bayern. In Jahrestagung der DVG-FG AVID; Kaiser Medien GmbH: Nürnberg, Germany, 2014. [Google Scholar]
- Moldal, T.; Vikøren, T.; Cliquet, F.; Marston, D.A.; van der Kooij, J.; Madslien, K.; Ørpetveit, I. First detection of European bat lyssavirus type 2 (EBLV-2) in Norway. BMC Vet. Res. 2017, 13, 216. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.; Goddard, T.M.; Goharriz, H.; Wise, E.; Jennings, D.; Selden, D.; Marston, D.A.; Banyard, A.C.; McElhinney, L.M.; Fooks, A.R. Two eblv-2 infected Daubenton’s bats detected in the north of England. Vet. Rec. 2016, 179, 311–312. [Google Scholar] [CrossRef] [PubMed]
- Nokireki, T.; Sironen, T.; Smura, T.; Karkamo, V.; Sihvonen, L.; Gadd, T. Second case of European bat lyssavirus type 2 detected in a Daubenton’s bat in Finland. Acta Vet. Scand. 2017, 59. [Google Scholar] [CrossRef] [PubMed]
- Hagner, N.; Ek-Kommonen, C.; Lokki, J.; Neuvonen, E.; Stjernberg, T.; Valle, M.; Veijalainen, P. No bat rabies found in Finland 1986. In European Bat Research; Hanák, V., Horácek, I., Gaisler, J., Eds.; Charles University Press: Praha, Czech Republic, 1989; pp. 631–635. [Google Scholar]
- Nokireki, T.; HuoVilainen, A.; Lilley, T.; Kyheroinen, E.M.; Ek-Kommonen, C.; Sihvonen, L.; Jakava-Viljanen, M. Bat rabies surveillance in Finland. BMC Vet. Res. 2013, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Van der Poel, W.H.; Van der Heide, R.; Verstraten, E.R.; Takumi, K.; Lina, P.H.; Kramps, J.A. European bat lyssaviruses, The Netherlands. Emerg. Infect. Dis. 2005, 11, 1854–1859. [Google Scholar] [CrossRef] [PubMed]
- Amengual, B.; Whitby, J.E.; King, A.; Cobo, J.S.; Bourhy, H. Evolution of European bat lyssaviruses. J. Gen. Virol. 1997, 78, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; McElhinney, L.M.; Johnson, N.; Muller, T.; Conzelmann, K.K.; Tordo, N.; Fooks, A.R. Comparative analysis of the full genome sequence of European bat lyssavirus type 1 and type 2 with other lyssaviruses and evidence for a conserved transcription termination and polyadenylation motif in the G-L 3′ non-translated region. J. Gen. Virol. 2007, 88, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.W.; Naisbitt, S.; Fan, J.S.; Sheng, M.; Zhang, M. The 8-kda dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J. Biol. Chem. 2001, 276, 14059–14066. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; McElhinney, L.M.; Ellis, R.J.; Horton, D.L.; Wise, E.L.; Leech, S.L.; David, D.; de Lamballerie, X.; Fooks, A.R. Next generation sequencing of viral RNA genomes. BMC Genom. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Brunker, K.; Marston, D.A.; Horton, D.L.; Cleaveland, S.; Fooks, A.R.; Kazwala, R.; Ngeleja, C.; Lembo, T.; Sambo, M.; Mtema, Z.J.; et al. Elucidating the phylodynamics of endemic rabies virus in Eastern Africa using whole-genome sequencing. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comParison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed]
- Stebbings, R.E.; Griffith, F. Distribution and Status of Bats in Europe; Institute of Terrestrial Ecology: Huntingdon, UK, 1986; p. 142. [Google Scholar]
- Smith, G.C.; Aegerter, J.N.; Allnutt, T.R.; MacNicoll, A.D.; Learmount, J.; Hutson, A.M.; Atterby, H. Bat population genetics and lyssavirus presence in Great Britain. Epidemiol. Infect. 2011, 139, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Aegerter, J.N.; Brookes, S.M.; McElhinney, L.M.; Jones, G.; Smith, G.C.; Fooks, A.R. Targeted surveillance for European bat lyssaviruses in English bats (2003–06). J. Wildl. Dis. 2009, 45, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Hutterer, R.; Ivanova, T.; Meyer-Cords, C.; Rodrigues, L. Bat migrations in Europe: A review of banding data and literature. Ger. Agency Nat. Conserv. 2005, 28, 83–84. [Google Scholar]
- Troupin, C.; Picard-Meyer, E.; Dellicour, S.; Casademont, I.; Kergoat, L.; Lepelletier, A.; Dacheux, L.; Baele, G.; Monchatre-Leroy, E.; Cliquet, F.; et al. Host genetic variation does not determine spatio-temporal patterns of European bat 1 lyssavirus. Genome Boil. Evol. 2017, 9, 3202–3213. [Google Scholar] [CrossRef] [PubMed]
- Marston, D.A.; Horton, D.L.; Nunez, J.; Ellis, R.J.; Orton, R.J.; Johnson, N.; Banyard, A.; McElhinney, L.; Freuling, C.; Firat, M.; et al. Genetic analysis of a rabies virus host shift event reveals within-host viral dynamics in a new host. Virus Evol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-scale phylogenomic analysis reveals the complex evolutionary history of rabies virus in multiple carnivore hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed]
- Hanke, D.; Freuling, C.M.; Fischer, S.; Hueffer, K.; Hundertmark, K.; Nadin-Davis, S.; Marston, D.; Fooks, A.R.; Botner, A.; Mettenleiter, T.C.; et al. Spatio-temporal analysis of the genetic diversity of arctic rabies viruses and their reservoir hosts in Greenland. PLoS Negl. Trop. Dis. 2016, 10, e0004779. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Original Reference | APHA Virus Ref. | PI Virus Ref. | Location | Country | Species | N400 Data Accession No. | Genome Accession No. | Case or Sequence Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1985 | FINMAN | RV8 | 9007FIN | Helsinki | Finland | Human | AY062091 | JX129233, KY688151 | [11] |
| 1987 | 41729 | RV30 | 9018HOL | Wommels | Netherlands | M.dasyc | EU293114 | EU293114 | [19] |
| 1987 | 47072 | RV29 | 9482HOL | Tjerkwerd | Netherlands | M.dasyc | U89480 | KY688145 | [25] |
| 1989 | 92666 | RV228 | 94112HOL | Andijk | Netherlands | M.dasyc | AY062089 | KY688146 | [19] |
| 1992 | TW1814/92 | RV594, RV2478 | - | Plaffeien | Switzerland | M.daub. | AY212117 | KY688133 | [26] |
| 1993 | - | - | 9375HOL | Roden | Netherlands | M.dasyc | AY863404 | KY688152 | [19] |
| 1993 | TW1392/93 | RV621, RV2479 | 9337SWI | Versoix | Switzerland | M.daub | AY212118 | KY688140 | [19] |
| 1996 | 18/96 | RV628 | EBL2GB | Sussex | England | M.daub | U89478 | KY688136 | [27] |
| 2002 | 105/02 | RV1332 | - | Lancashire | England | M.daub | AY212120 | KY688150 | [28] |
| 2002 | Human case | RV1333 | - | Angus | Scotland | Human | EF157977 | EF157977 | [29] |
| 2002 | TW0118/02 | RV2480 | 02053SWI | Geneva | Switzerland | M.daub | AY863408 | KY688132 | [19] |
| 2004 | 603/04 | RV1787 | - | Surrey | England | M.daub | JQ796807 | KY688142 | [30] |
| 2003 | 696/04 | RV1788 | - | Lancashire | England | M.daub | JQ796808 | KF155004 | [31] |
| 2006 | 672/06 | RV2159 | - | Oxfordshire | England | M.daub | JQ796809 | KY688144 | [32] |
| 2006 | 18856 | RV2506 | - | Magdeburg | Germany | M.daub | JQ796805 | KY688135 | [33] |
| 2006 | 25538 | - | - | Thuringia | Germany | M.daub | KF826115 | KY688137 | [34] |
| 2007 | 16618 | RV2505 | - | Bad Buchau | Germany | M.daub | GU227648 | KY688138 | [35] |
| 2007 | 762/07 | RV2336 | - | Shropshire | England | M.daub | JQ796810 | KY688139 | [36] |
| 2008 | 166/08 | RV2418 | - | Surrey | England | M.daub | JQ796811 | KY688141 | [37] |
| 2008 | 1218/08 | RV2473 | - | Shropshire | England | M.daub | JQ796812 | KY688147 | [38] |
| 2009 | 882/09 | RV2482 | - | West Lothian | Scotland | M.daub | JQ796806 | KY688143 | [39] |
| 2009 | ra3278/09 | - | - | Nr Turku | Finland | M.daub | GU002399 | JX129232 | [40] |
| 2012 | F500/LGL | - | - | Ingolstadt, Bavaria | Germany | M.daub | - | KY688149 | [34,41] |
| 2013 | 31452 | - | - | Gießen, Hesse | Germany | M.daub | KF826149 | KY688134 | [34] |
| 2013 | F860/LGL | - | - | Ingolstadt, Bavaria | Germany | M.daub | - | Not available | This study |
| 2014 | 281/14 | RV2974 | - | Shropshire | England | M.daub | - | KY688148 | This study |
| 2015 | 421/15 | RV3158 | - | Newtown, Powys | Wales | M.daub | - | KY688153 | This study |
| 2015 | 2015-04-18832 | - | - | Oppland | Norway | M.daub | - | KY688154 | [42] |
| 2016 | 307/16 | RV3369 | - | North Yorkshire | England | M.daub | - | KY688156 | This study |
| 2016 | 432/16 | RV3370 | - | Northumberland | England | M.daub | - | KY688155 | [43] |
| 2016 | 2Finland2016 | - | - | Inkoo, Uusimaa | Finland | M.daub | MF326269 | Not available | [44] |
| 2017 | 591/17 | RV3385 | - | Derbyshire | England | M.daub | - | Not available | This study |
| Active Surveillance Detection of Viral RNA in Oro-Pharyngeal Swab Only: | |||||||||
| 2008 | 80063 | M08/09 | - | Perthshire | Scotland | M.daub | JQ796804 | Not available | [20] |
| 2009 | 70 | - | - | Genthod | Switzerland | M.daub | HM067110 | Not available | [26] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McElhinney, L.M.; Marston, D.A.; Wise, E.L.; Freuling, C.M.; Bourhy, H.; Zanoni, R.; Moldal, T.; Kooi, E.A.; Neubauer-Juric, A.; Nokireki, T.; et al. Molecular Epidemiology and Evolution of European Bat Lyssavirus 2. Int. J. Mol. Sci. 2018, 19, 156. https://doi.org/10.3390/ijms19010156
McElhinney LM, Marston DA, Wise EL, Freuling CM, Bourhy H, Zanoni R, Moldal T, Kooi EA, Neubauer-Juric A, Nokireki T, et al. Molecular Epidemiology and Evolution of European Bat Lyssavirus 2. International Journal of Molecular Sciences. 2018; 19(1):156. https://doi.org/10.3390/ijms19010156
Chicago/Turabian StyleMcElhinney, Lorraine M., Denise A. Marston, Emma L. Wise, Conrad M. Freuling, Hervé Bourhy, Reto Zanoni, Torfinn Moldal, Engbert A. Kooi, Antonie Neubauer-Juric, Tiina Nokireki, and et al. 2018. "Molecular Epidemiology and Evolution of European Bat Lyssavirus 2" International Journal of Molecular Sciences 19, no. 1: 156. https://doi.org/10.3390/ijms19010156