Influence of Lytic Polysaccharide Monooxygenase Active Site Segments on Activity and Affinity

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
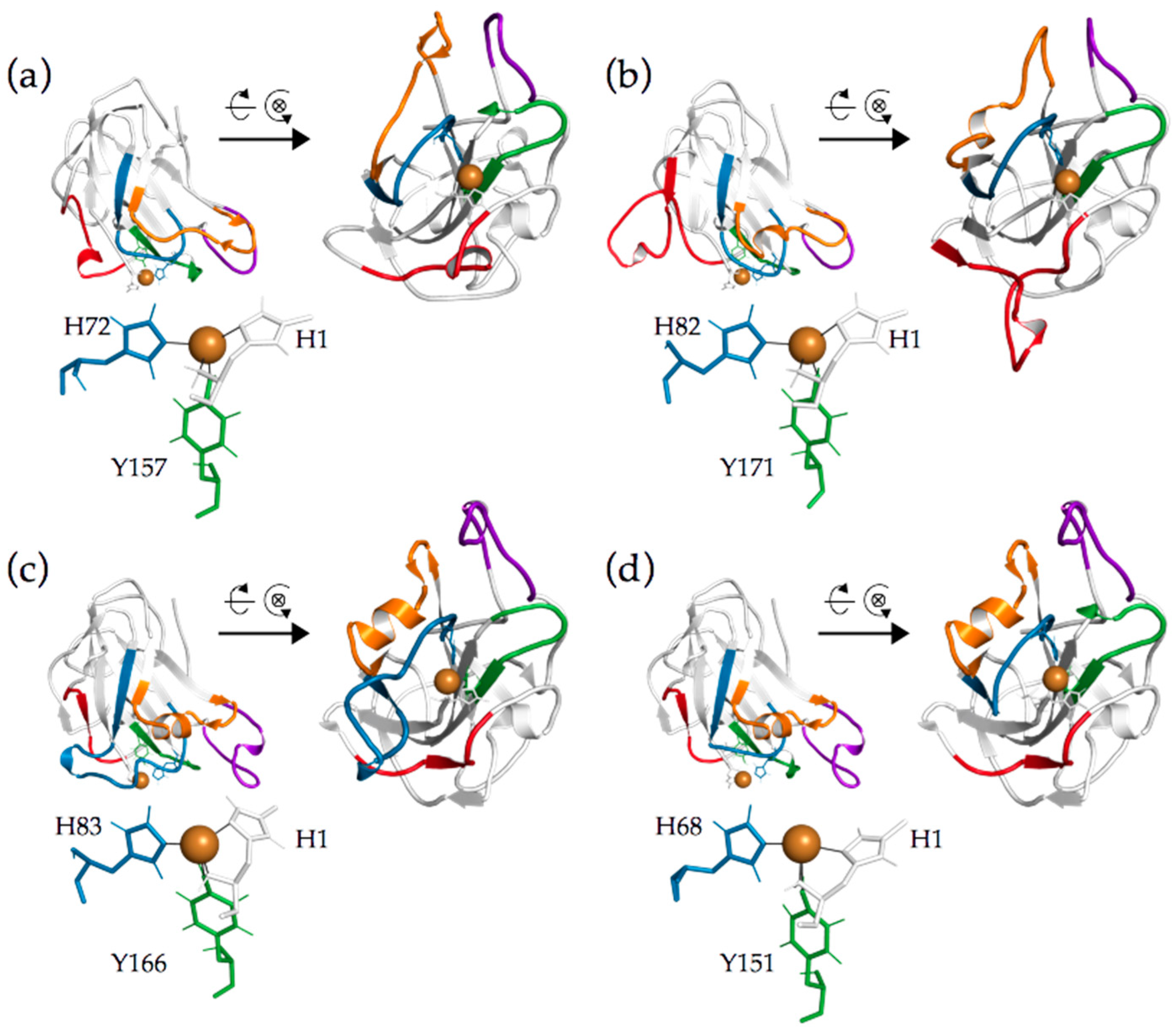
2.1. Clustering of AA9 LPMOs Is Based on Extended Catalytic Site Segments
2.2. Analysis of Active Site Segments Hints Towards Regioselectivity
2.3. Thermal Stability of Purified LPMOs
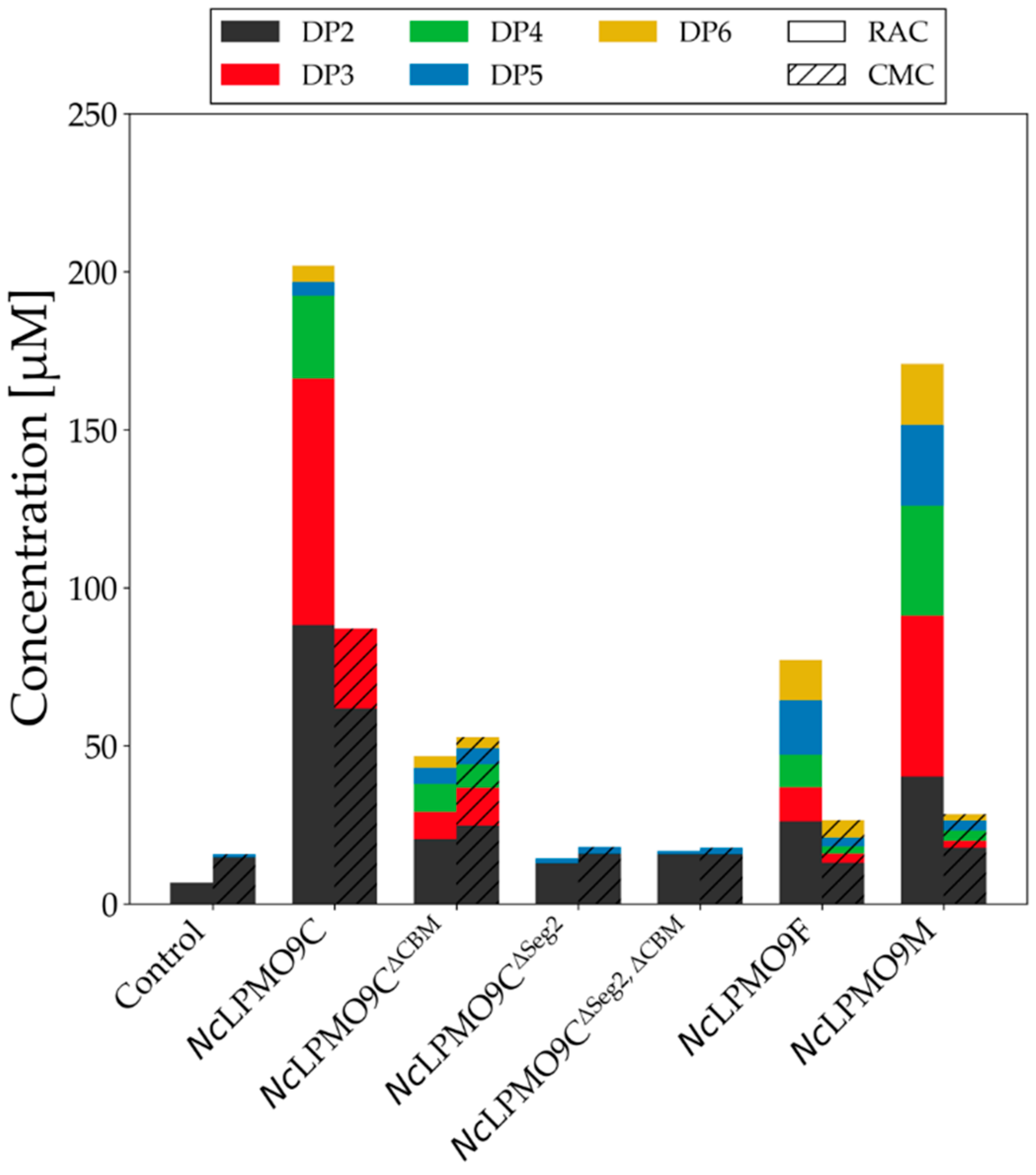
2.4. LPMO Activity on Cellulosic Substrates
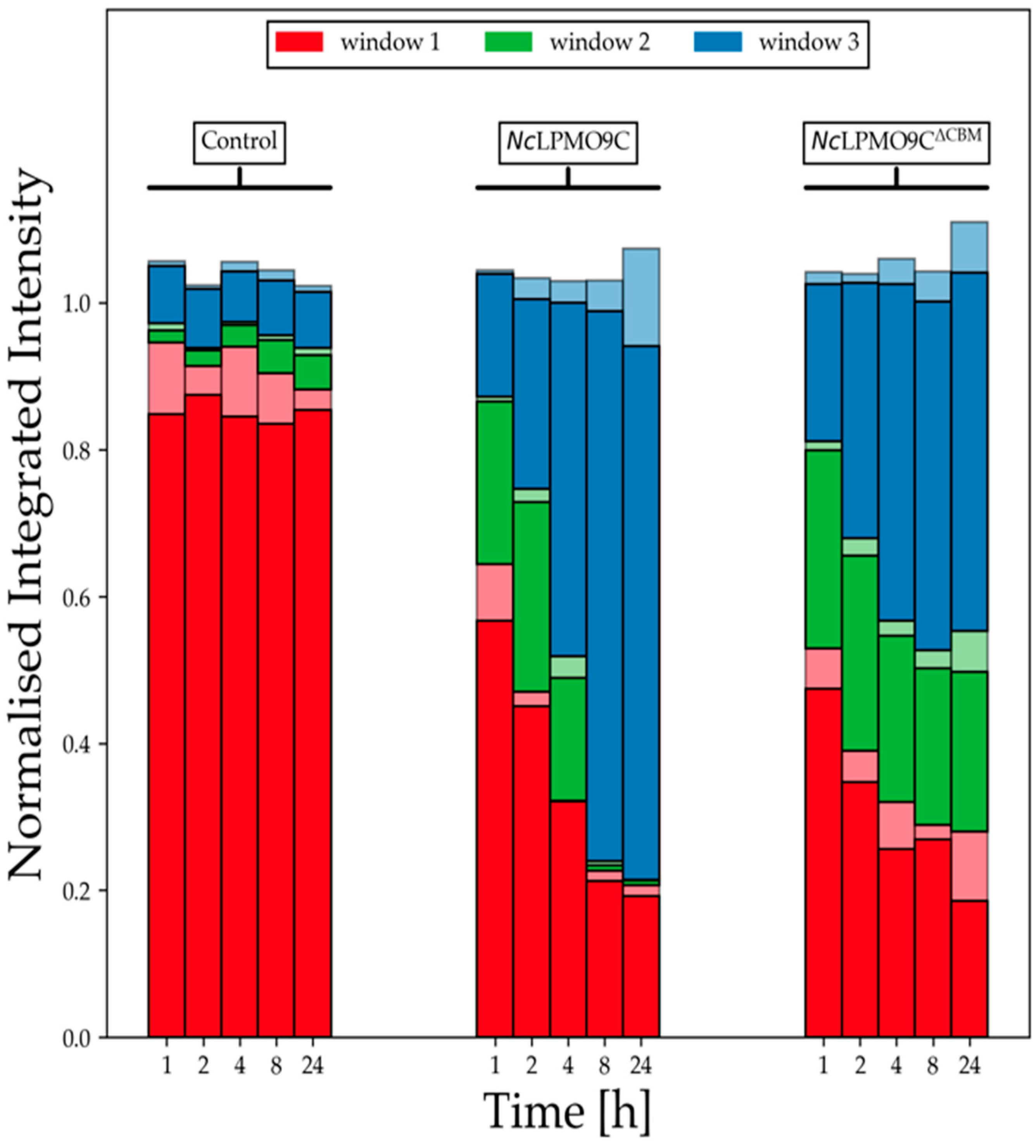
2.5. LPMO Activity on Xyloglucan
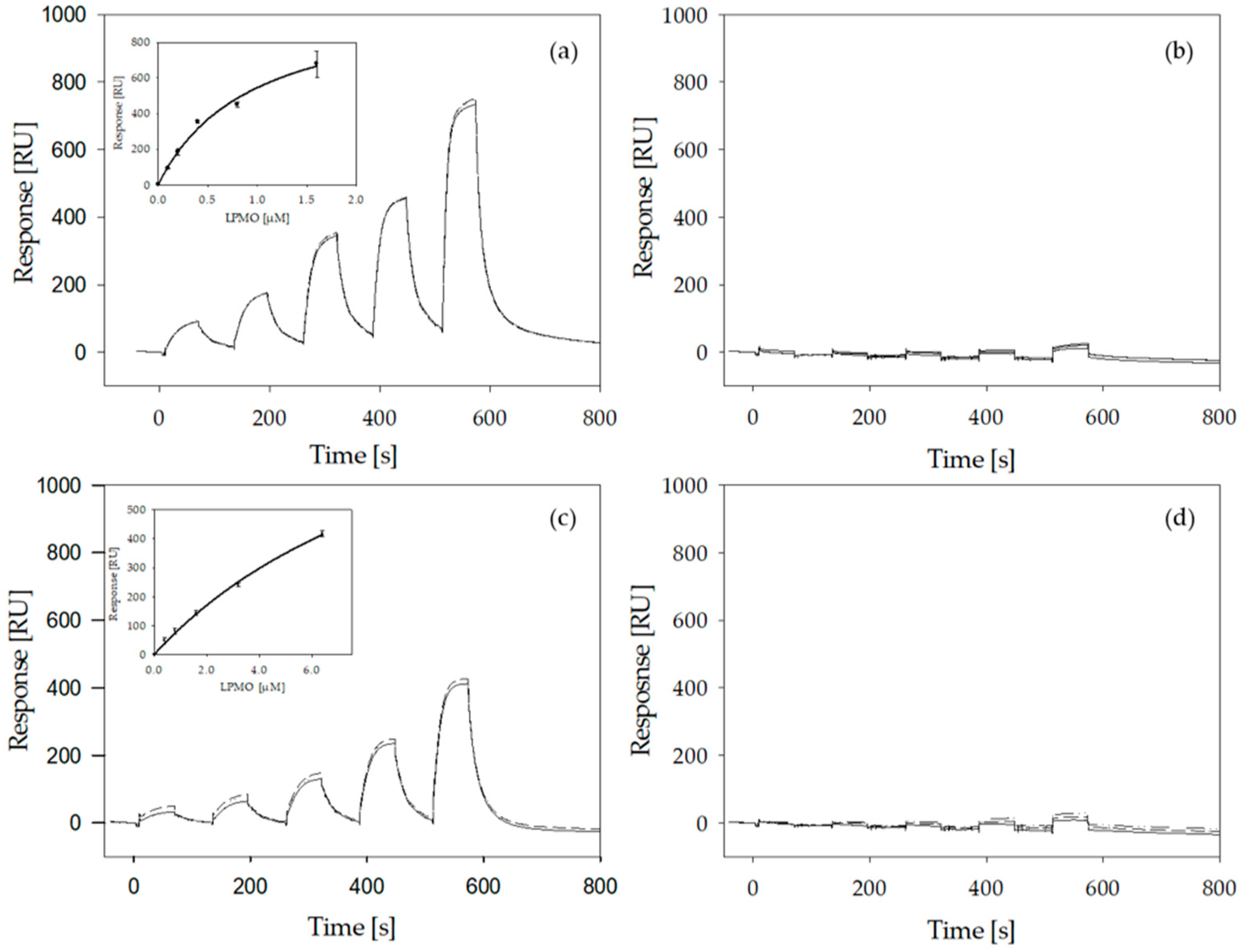
2.6. LPMO Binding to Cellulose
3. Discussion
4. Materials and Methods
4.1. Multiple Sequence Alignment, Phylogenetic Analysis and Homology Modelling
4.2. Chemicals and Substrates
4.3. Plasmid Design
4.4. Enzyme Production and Purification
4.5. Protein Identification: Peptide Mapping Analysis
4.6. LPMO Activity Assay
4.7. Enzyme Incubations
4.8. Differential Scanning Calorimetry
4.9. Electronic Circular Dichroism (ECD) Spectroscopy
4.10. HPAEC Analysis for Profiling Oligosaccharides
4.11. HPSEC Analysis for Molecular Weight Distribution of (Degraded) Xyloglucan
4.12. Binding Studies on Cellulose
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Auxiliary activity enzyme |
| CAYZy | Carbohydrate-active enzyme |
| CBM1 | Carbohydrate binding module family 1 |
| CMC | Carboxymethyl cellulose |
| DP | Degree of polymerization |
| DSC | Differential scanning calorimetry |
| ECD | Electronic circular dichroism |
| HPAEC-PAD | High performance anion exchange chromatography coupled to a pulsed amperometric detector |
| HPSEC-RI | High performance size exclusion chromatography coupled to a refractive index detector |
| LC-ESI-MS | Liquid chromatography-electrospray ionization mass spectrometry |
| LPMO | Lytic polysaccharide monooxygenase |
| RAC | Regenerated amorphous cellulose |
| SPR | Surface plasmon resonance |
| XG | Tamarind seed xyloglucan |
References
- Vaaje-Kolstad, G.; Westereng, B.; Horn, S.J.; Liu, Z.; Zhai, H.; Sørlie, M.; Eijsink, V.G.H. An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 2010, 330, 219–222. [Google Scholar] [CrossRef]
- Harris, P.V.; Welner, D.; McFarland, K.C.; Re, E.; Poulsen, J.-C.N.; Brown, K.; Salbo, R.; Ding, H.; Vlasenko, E.; Merino, S.; et al. Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: Structure and function of a large, enigmatic family. Biochemistry 2010, 49, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.-C.; Kracher, D.; Gandini, R.; Sygmund, C.; Kittl, R.; Haltrich, D.; Hällberg, B.M.; Ludwig, R.; Divne, C. Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat. Commun. 2015, 6, 7542. [Google Scholar] [CrossRef] [Green Version]
- Beeson, W.T.; Phillips, C.M.; Cate, J.H.D.; Marletta, M.A. Oxidative cleavage of cellulose by fungal copper-dependent polysaccharide monooxygenases. J. Am. Chem. Soc. 2012, 134, 890–892. [Google Scholar] [CrossRef] [PubMed]
- Hangasky, J.A.; Iavarone, A.T.; Marletta, M.A. Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2018, 115, 4915–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, R.J.; Sweeney, M.D.; Lo Leggio, L.; Otten, H.; Poulsen, J.-C.N.; Johansen, K.S.; Krogh, K.B.R.M.; Jørgensen, C.I.; Tovborg, M.; Anthonsen, A.; et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. USA 2011, 108, 15079–15084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannella, D.; Möllers, K.B.B.; Frigaard, N.-U.; Jensen, P.E.E.; Bjerrum, M.J.J.; Johansen, K.S.S.; Felby, C. Light-driven oxidation of polysaccharides by photosynthetic pigments and a metalloenzyme. Nat. Commun. 2016, 7, 11134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmayr, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 2016, 352, 1098. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Levasseur, A.; Drula, E.; Lombard, V.; Coutinho, P.M.; Henrissat, B. Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol. Biofuels 2013, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Hemsworth, G.R.; Henrissat, B.; Davies, G.J.; Walton, P.H. Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol. 2014, 10, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Vu, V.V.; Beeson, W.T.; Span, E.A.; Farquhar, E.R.; Marletta, M.A. A family of starch-active polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2014, 111, 13822–13827. [Google Scholar] [CrossRef] [Green Version]
- Couturier, M.; Ladevèze, S.; Sulzenbacher, G.; Ciano, L.; Fanuel, M.; Moreau, C.; Villares, A.; Cathala, B.; Chaspoul, F.; Frandsen, K.E.; et al. Lytic xylan oxidases from wood-decay fungi unlock biomass degradation. Nat. Chem. Biol. 2018, 14, 306–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbadin, F.; Hemsworth, G.R.; Ciano, L.; Henrissat, B.; Dupree, P.; Tryfona, T.; Marques, R.D.S.; Sweeney, S.T.; Besser, K.; Elias, L.; et al. An ancient family of lytic polysaccharide monooxygenases with roles in arthropod development and biomass digestion. Nat. Commun. 2018, 9, 756. [Google Scholar] [CrossRef]
- Filiatrault-Chastel, C.; Navarro, D.; Haon, M.; Grisel, S.; Herpoël-Gimbert, I.; Chevret, D.; Fanuel, M.; Henrissat, B.; Heiss-Blanquet, S.; Margeot, A.; et al. AA16, a new lytic polysaccharide monooxygenase family identified in fungal secretomes. Biotechnol. Biofuels 2019, 12, 55. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Røhr, Å.K.; Müller, G.; Chylenski, P.; Skaugen, M.; Forsberg, Z.; Horn, S.J.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol. 2017, 13, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Chylenski, P.; Bissaro, B.; Sørlie, M.; Røhr, Å.K.; Várnai, A.; Horn, S.J.; Eijsink, V.G.H. Lytic polysaccharide monooxygenases in enzymatic processing of lignocellulosic biomass. ACS Catal. 2019, 9, 4970–4991. [Google Scholar] [CrossRef]
- Vaaje-Kolstad, G.; Houston, D.R.; Riemen, A.H.K.; Eijsink, V.G.H.; van Aalten, D.M.F. Crystal structure and binding properties of the Serratia marcescens chitin-binding protein CBP21. J. Biol. Chem. 2005, 280, 11313–11319. [Google Scholar] [CrossRef] [Green Version]
- Vu, V.V.; Beeson, W.T.; Phillips, C.M.; Cate, J.H.D.; Marletta, M.A. Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 2014, 136, 562–565. [Google Scholar] [CrossRef]
- Li, X.; Beeson IV, W.T.; Phillips, C.M.; Marletta, M.A.; Cate, J.H.D. Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 2012, 20, 1051–1061. [Google Scholar] [CrossRef] [Green Version]
- Patel, I.; Kracher, D.; Ma, S.; Garajova, S.; Haon, M.; Faulds, C.B.; Berrin, J.-G.; Ludwig, R.; Record, E. Salt-responsive lytic polysaccharide monooxygenases from the mangrove fungus Pestalotiopsis sp. NCi6. Biotechnol. Biofuels 2016, 9, 108. [Google Scholar] [CrossRef]
- Lenfant, N.; Hainaut, M.; Terrapon, N.; Drula, E.; Lombard, V.; Henrissat, B. A bioinformatics analysis of 3400 lytic polysaccharide oxidases from family AA9. Carbohydr. Res. 2017, 448, 166–174. [Google Scholar] [CrossRef]
- Wu, M.; Beckham, G.T.; Larsson, A.M.; Ishida, T.; Kim, S.; Payne, C.M.; Himmel, M.E.; Crowley, M.F.; Horn, S.J.; Westereng, B.; et al. Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the basidiomycota fungus Phanerochaete chrysosporium. J. Biol. Chem. 2013, 288, 12828–12839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisova, A.S.; Isaksen, T.; Dimarogona, M.; Kognole, A.A.; Mathiesen, G.; Várnai, A.; Røhr, Å.K.; Payne, C.M.; Sørlie, M.; Sandgren, M.; et al. Structural and functional characterization of a lytic polysaccharide monooxygenase with broad substrate specificity. J. Biol. Chem. 2015, 290, 22955–22969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danneels, B.; Tanghe, M.; Joosten, H.J.; Gundinger, T.; Spadiut, O.; Stals, I.; Desmet, T. A quantitative indicator diagram for lytic polysaccharide monooxygenases reveals the role of aromatic surface residues in HjLPMO9A regioselectivity. PLoS ONE 2017, 12, e0178446. [Google Scholar] [CrossRef] [PubMed]
- Courtade, G.; Forsberg, Z.; Heggset, E.B.; Eijsink, V.G.H.; Aachmann, F.L. The carbohydrate-binding module and linker of a modular lytic polysaccharide monooxygenase promote localized cellulose oxidation. J. Biol. Chem. 2018, 293, 13006–13015. [Google Scholar] [CrossRef] [Green Version]
- Danneels, B.; Tanghe, M.; Desmet, T. Structural features on the dubstrate-binding surface of fungal lytic polysaccharide monooxygenases determine their oxidative regioselectivity. Biotechnol. J. 2019, 14, 1800211. [Google Scholar] [CrossRef]
- Forsberg, Z.; Bissaro, B.; Gullesen, J.; Dalhus, B.; Vaaje-Kolstad, G.; Eijsink, V.G.H. Structural determinants of bacterial lytic polysaccharide monooxygenase functionality. J. Biol. Chem. 2018, 293, 1397–1412. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.M.; Beeson, W.T.; Cate, J.H.; Marletta, M.A. Cellobiose dehydrogenase and a copper-dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem. Biol. 2011, 6, 1399–1406. [Google Scholar] [CrossRef]
- Bennati-Granier, C.; Garajova, S.; Champion, C.; Grisel, S.; Haon, M.; Zhou, S.; Fanuel, M.; Ropartz, D.; Rogniaux, H.; Gimbert, I.; et al. Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 2015, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Jagadeeswaran, G.; Gainey, L.; Mort, A.J. An AA9-LPMO containing a CBM1 domain in Aspergillus nidulans is active on cellulose and cleaves cello-oligosaccharides. AMB Express 2018, 8, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalak, A.; Villares, A.; Moreau, C.; Haon, M.; Grisel, S.; D’Orlando, A.; Herpoël-Gimbert, I.; Labourel, A.; Cathala, B.; Berrin, J.-G. Influence of the carbohydrate-binding module on the activity of a fungal AA9 lytic polysaccharide monooxygenase on cellulosic substrates. Biotechnol. Biofuels 2019, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommhagen, M.; Westphal, A.H.; van Berkel, W.J.H.; Kabel, M.A. Distinct substrate specificities and electron-donating systems of fungal lytic polysaccharide monooxygenases. Front. Microbiol. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Eichenberger, A.P.; Allison, J.R.; Dolenc, J.; Geerke, D.P.; Horta, B.A.C.; Meier, K.; Oostenbrink, C.; Schmid, N.; Steiner, D.; Wang, D.; et al. GROMOS++ software for the analysis of biomolecular simulation trajectories. J. Chem. Theory Comput. 2011, 7, 3379–3390. [Google Scholar] [CrossRef] [PubMed]
- Breslmayr, E.; Hanžek, M.; Hanrahan, A.; Leitner, C.; Kittl, R.; Šantek, B.; Oostenbrink, C.; Ludwig, R. A fast and sensitive activity assay for lytic polysaccharide monooxygenase. Biotechnol. Biofuels 2018, 11, 79. [Google Scholar] [CrossRef] [PubMed]
- Kittl, R.; Kracher, D.; Burgstaller, D.; Haltrich, D.; Ludwig, R. Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 2012, 5, 79. [Google Scholar] [CrossRef] [Green Version]
- Durowoju, I.B.; Bhandal, K.S.; Hu, J.; Carpick, B.; Kirkitadze, M. Differential scanning calorimetry—A method for assessing the thermal stability and conformation of protein antigen. J. Vis. Exp. 2017, 121, e55262. [Google Scholar]
- Westereng, B.; Ishida, T.; Vaaje-Kolstad, G.; Wu, M.; Eijsink, V.G.H.; Igarashi, K.; Samejima, M.; Ståhlberg, J.; Horn, S.J.; Sandgren, M. The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal-dependent oxidative enzyme that cleaves cellulose. PLoS ONE 2011, 6, e27807. [Google Scholar] [CrossRef] [Green Version]
- Forsberg, Z.; Vaaje-Kolstad, G.; Westereng, B.; Bunaes, A.C.; Stenstrøm, Y.; MacKenzie, A.; Sørlie, M.; Horn, S.J.; Eijsink, V.G.H. Cleavage of cellulose by a CBM33 protein. Protein Sci. 2011, 20, 1479–1483. [Google Scholar] [CrossRef] [Green Version]
- Frommhagen, M.; Koetsier, M.J.; Westphal, A.H.; Visser, J.; Hinz, S.W.A.; Vincken, J.P.; Van Berkel, W.J.H.; Kabel, M.A.; Gruppen, H. Lytic polysaccharide monooxygenases from Myceliophthora thermophila C1 differ in substrate preference and reducing agent specificity. Biotechnol. Biofuels 2016, 9, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agger, J.W.; Isaksen, T.; Varnai, A.; Vidal-Melgosa, S.; Willats, W.G.T.; Ludwig, R.; Horn, S.J.; Eijsink, V.G.H.; Westereng, B.; Várnai, A.; et al. Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc. Natl. Acad. Sci. USA 2014, 111, 6287–6292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bey, M.; Zhou, S.; Poidevin, L.; Henrissat, B.; Coutinho, P.M.; Berrin, J.-G.G.; Sigoillot, J.-C.C. Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl. Environ. Microbiol. 2013, 79, 488–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanghe, M.; Danneels, B.; Last, M.; Beerens, K.; Stals, I.; Desmet, T. Disulfide bridges as essential elements for the thermostability of lytic polysaccharide monooxygenase LPMO10C from Streptomyces coelicolor. Protein Eng. Des. Sel. 2017, 30, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Blossom, B.M.; Russo, D.A.; van Oort, B.; Croce, R.; Jensen, P.E.; Felby, C.; Bjerrum, M.J. Thermal unfolding and refolding of a lytic polysaccharide monooxygenase from Thermoascus aurantiacus. RSC Adv. 2019, 9, 29734–29742. [Google Scholar] [CrossRef] [Green Version]
- Kracher, D.; Andlar, M.; Furtmüller, P.G.; Ludwig, R. Active-site copper reduction promotes substrate binding of fungal lytic polysaccharide monooxygenase and reduces stability. J. Biol. Chem. 2018, 293, 1676–1687. [Google Scholar] [CrossRef] [Green Version]
- Frommhagen, M.; Westphal, A.H.; Hilgers, R.; Koetsier, M.J.; Hinz, S.W.A.; Visser, J.; Gruppen, H.; van Berkel, W.J.H.; Kabel, M.A. Quantification of the catalytic performance of C1-cellulose-specific lytic polysaccharide monooxygenases. Appl. Microbiol. Biotechnol. 2018, 102, 1281–1295. [Google Scholar] [CrossRef] [Green Version]
- Rodger, A.; Ismail, M.A. Introduction to circular dichroism (Chapter 4). In Spectrophotometry and Spectrofluorimentry; Gore, M.G., Ed.; Oxford University Press: Avon, UK, 2000; pp. 99–139. ISBN 978-0199638130. [Google Scholar]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.P.; Moret, B.M.E.; Stamatakis, A. How many bootstrap replicates are necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, M.; Kiefer, F.; Biasini, M.; Bordoli, L.; Schwede, T. Modeling protein quaternary structure of homo- and hetero-oligomers beyond binary interactions by homology. Sci. Rep. 2017, 7, 10480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30, S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.P.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-H.P.; Cui, J.; Lynd, L.R.; Kuang, L.R. A transition from cellulose swelling to cellulose dissolution by o-phosphoric acid: Evidence from enzymatic hydrolysis and supramolecular structure. Biomacromolecules 2006, 7, 644–648. [Google Scholar] [CrossRef]
- Micsonai, A.; Wien, F.; Bulyáki, É.; Kun, J.; Moussong, É.; Lee, Y.-H.; Goto, Y.; Réfrégiers, M.; Kardos, J. BeStSel: A web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. [Google Scholar] [CrossRef]
- Raj, G.; Balnois, E.; Baley, C.; Grohens, Y. Probing interfacial interactions in natural fibre reinforced biocomposites using colloidal force microscopy. In Natural Polymers. Volume 1: Composites; John, M.J., Sabu, T., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2012; pp. 183–203. ISBN 978-1-84973-402-8. [Google Scholar]
- Sczech, R.; Riegler, H. Molecularly smooth cellulose surfaces for adhesion studies. J. Colloid Interface Sci. 2006, 301, 376–385. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | KM [µM] | Spec. act. with 100 µM H2O2 [U g−1] | kcat [s−1] | kcat/KM [µM−1 s−1] |
|---|---|---|---|---|
| NcLPMO9C | 2.8 ± 0.1 | 28.8 ± 0.2 | 0.57 ± 0.03 | 0.20 ± 0.02 |
| NcLPMO9C∆CBM | 1.4 ± 0.1 | 13.0 ± 0.1 | 0.39 ± 0.01 | 0.28 ± 0.03 |
| NcLPMO9C∆Seg2 | 5.4 ± 0.1 | 42.4 ± 0.2 | 0.55 ± 0.01 | 0.10 ± 0.01 |
| NcLPMO9C∆Seg2, ∆CBM | 4.0 ± 0.1 | 41.1 ± 0.3 | 0.37 ± 0.01 | 0.09 ± 0.01 |
| NcLPMO9M | 7.0 ± 0.3 | 39.1 ± 0.3 | 0.40 ± 0.01 | 0.06 ± 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laurent, C.V.F.P.; Sun, P.; Scheiblbrandner, S.; Csarman, F.; Cannazza, P.; Frommhagen, M.; van Berkel, W.J.H.; Oostenbrink, C.; Kabel, M.A.; Ludwig, R. Influence of Lytic Polysaccharide Monooxygenase Active Site Segments on Activity and Affinity. Int. J. Mol. Sci. 2019, 20, 6219. https://doi.org/10.3390/ijms20246219
Laurent CVFP, Sun P, Scheiblbrandner S, Csarman F, Cannazza P, Frommhagen M, van Berkel WJH, Oostenbrink C, Kabel MA, Ludwig R. Influence of Lytic Polysaccharide Monooxygenase Active Site Segments on Activity and Affinity. International Journal of Molecular Sciences. 2019; 20(24):6219. https://doi.org/10.3390/ijms20246219
Chicago/Turabian StyleLaurent, Christophe V.F.P., Peicheng Sun, Stefan Scheiblbrandner, Florian Csarman, Pietro Cannazza, Matthias Frommhagen, Willem J.H. van Berkel, Chris Oostenbrink, Mirjam A. Kabel, and Roland Ludwig. 2019. "Influence of Lytic Polysaccharide Monooxygenase Active Site Segments on Activity and Affinity" International Journal of Molecular Sciences 20, no. 24: 6219. https://doi.org/10.3390/ijms20246219