Combinations of Spok genes create multiple meiotic drivers in Podospora
- Uppsala University, Sweden
- University of Bordeaux, France
- Wageningen University, Netherlands
Abstract
Meiotic drive is the preferential transmission of a particular allele during sexual reproduction. The phenomenon is observed as spore killing in multiple fungi. In natural populations of Podospora anserina, seven spore killer types (Psks) have been identified through classical genetic analyses. Here we show that the Spok gene family underlies the Psks. The combination of Spok genes at different chromosomal locations defines the spore killer types and creates a killing hierarchy within a population. We identify two novel Spok homologs located within a large (74–167 kbp) region (the Spok block) that resides in different chromosomal locations in different strains. We confirm that the SPOK protein performs both killing and resistance functions and show that these activities are dependent on distinct domains, a predicted nuclease and kinase domain. Genomic and phylogenetic analyses across ascomycetes suggest that the Spok genes disperse through cross-species transfer, and evolve by duplication and diversification within lineages.
https://doi.org/10.7554/eLife.46454.001eLife digest
In many organisms, most cells carry two versions of a given gene, one coming from the mother and the other from the father. An exception is sexual cells such as eggs, sperm, pollen or spores, which should only contain one variant of a gene. During their formation, these cells usually have an equal chance of inheriting one of the two gene versions.
However, a certain class of gene variants called meiotic drivers can cheat this process and end up in more than half of the sexual cells; often, the cells that contain the drivers can kill sibling cells that do not carry these variants. This results in the selfish genetic elements spreading through populations at a higher rate, sometimes with severe consequences such as shifting the ratio of males to females.
Meiotic drivers have been discovered in a wide range of organisms, from corn to mice to fruit flies and bread mold. They also exist in the fungus Podospora anserina, where they are called ‘spore killers’. Fungi are often used to study complex genetic processes, yet the identity and mode of action of spore killers in P. anserina were still unknown.
Vogan, Ament-Velásquez et al. used a combination of genetic methods to identify three genes from the Spok family which are responsible for certain spores being able to kill their siblings. Two of these were previously unknown, and they could be found in different locations throughout the genome as part of a larger genetic region. Depending on the combination of Spok genes it carries, a spore can kill or be protected against other spores that contain different permutations of the genes. Copies of these genes were also shown to be present in other fungi, including species that are a threat to crops.
Scientists have already started to create synthetic meiotic drivers to manipulate how certain traits are inherited within a population. This could be useful to control or eradicate pests and insects that transmit dangerous diseases. The results by Vogan, Ament-Velásquez et al. shine a light on the complex ways that natural meiotic drivers work, including how they can be shared between species; this knowledge could inform how to safely deploy synthetic drivers in the wild.
https://doi.org/10.7554/eLife.46454.002Introduction
The genomes of all Eukaryotes harbor selfish genetic elements that employ a variety of mechanisms to undermine the canonical modes of DNA replication and meiosis in order to bias their own transmission (Werren et al., 1988; Burt and Trivers, 2009). As the proliferation of these elements is independent of the regulated reproduction of the host organism, they can create conflict within the genome (Rice and Holland, 1997). Such intragenomic conflict is predicted by theory to spur an arms race between the genome and the elements and, consequently, to act as a major driver of evolutionary change (Werren, 2011). To understand the extent to which intragenomic conflict has shaped the evolution of genomes and populations, it is crucial to identify the selfish genetic elements that are able to impact the dynamics of natural populations.
One important class of selfish genetic elements are known as meiotic drivers. These elements use a variety of mechanisms to hijack meiosis in order to bias their transmission to the gametes in proportions greater than 50% (Sandler and Novitski, 1957). This segregation distortion of alleles can be difficult to observe unless it is linked to an obvious phenotype such as sex (Sandler and Novitski, 1957; Helleu et al., 2014), thus the prevalence of meiotic drive in nature is probably underestimated. Nevertheless, meiotic drive has been observed in many model systems, including Drosophila, Mus, Neurospora, and Zea mays, suggesting that it is widespread across all major Eukaryotic groups (Lindholm et al., 2016; Bravo Núñez et al., 2018b). In ascomycete fungi, meiotic drive occurs in the form of spore killing, which represents the most direct way to observe the presence of drive (Turner and Perkins, 1991). When a strain possessing a driving allele mates with a compatible strain that does not carry the allele (i.e., a sensitive strain), the meiotic products (ascospores) that carry the driving allele will induce the abortion of their sibling spores that do not have the allele. Spore killing is apparent in the sexual structures (asci) of the fungi because it results in half of the normal number of viable spores. Owing to the haplontic life cycle of most fungi, spore killing is unusual among meiotic drivers as it is the only system in which the offspring of an organism are killed by the drive (Lyttle, 1991). In addition, with few exceptions (Hammond et al., 2012; Svedberg et al., 2018), spore killer elements appear to be governed by single loci that confer both killing and resistance (Grognet et al., 2014; Nuckolls et al., 2017; Hu et al., 2017), which contrasts with the other well-studied drive systems that comprise genomic regions as large as entire chromosomes (Larracuente and Presgraves, 2012; Hammer et al., 1989).
Meiotic drivers are often expected to reach fixation or extinction in populations relatively rapidly (Crow, 1991), at which point the effects of the drivers will no longer be observable. In agreement with this expectation, most drivers that have been described exhibit large shifts in frequencies in both time and space (Lindholm et al., 2016; Carvalho and Vaz, 1999). In the case of spore killers, multiple drivers have been found to coexist within a given species. The evolutionary dynamics of multiple drivers within species has not been thoroughly explored, but two contrasting examples are known. In the genomes of Schizosaccharomyces pombe, numerous copies of both functional and pseudogenized versions of the wtf driver genes are found (Nuckolls et al., 2017; Hu et al., 2017; Eickbush et al., 2019). By contrast, the two spore killers Sk-2 and Sk-3 of Neurospora intermedia have only been described in wild strains four times and once, respectively, whereas resistance to spore killing is widespread (Turner, 2001). The impact of multiple drivers coexisting in a single population was not characterized in either of these cases.
Natural populations of the filamentous fungus Podospora anserina are known to host multiple spore killers (Grognet et al., 2014; van der Gaag et al., 2000; Hamann and Osiewacz, 2004), and hence provide an ideal system for the investigation of interactions among drivers at the population level. The first spore killer gene to be described in P. anserina was het-s, a gene that is also involved in allorecognition (Dalstra et al., 2003). Another class of spore killer genes in Podospora are known as Spok genes. Spok1 is only known from a single representative of P. comata, a species that is closely related to P. anserina, whereas Spok2 has been shown to exist in high frequency among strains of a French population of P. anserina (Grognet et al., 2014). Spok1 is capable of killing in the presence of Spok2, but not vice versa, indicating a dominant epistatic relationship between the two genes. In addition, seven spore killer genotypes have been identified through classical genetic analysis (van der Gaag et al., 2000). These are referred to as Psk-1 through Psk-7 and were defined by observing the presence, absence and frequency of killed spores in defined crosses among French and Dutch P. anserina strains (Box 1—figure 1). At the onset of this study, it was not known whether the Psk elements represent independent meiotic drive genes, or whether they may be related to the Spoks and/or allorecognition loci. The het-s gene itself is not associated with the Psks, but allorecognition is correlated with Psk spore killing (van der Gaag et al., 2003). On the other hand, the relationship between the Spoks is reminiscent of the hierarchy of killing among the Psks, suggesting a possible connection between the activity of Spok genes and Psks.
Box 1.
The seven separate Psks are defined by their spore-killing percentage and mutual interactions.
To understand how the spore-killing percentages relate to the Psk types of the strains, it is necessary to first appreciate some of the fundamental aspects of Podospora biology. Within the fruiting body (perithecium), dikaryotic cells undergo karyogamy to produce a diploid nucleus that immediately enters meiosis. After meiosis, one round of post-meiotic mitosis occurs, resulting in eight daughter nuclei. The nuclei are packaged together with their non-sister nuclei from mitosis (dashed line) to generate dikaryotic, self-compatible spores. In a cross in which the parental strains harbor two alternative alleles for a given gene of interest (one of which is indicated by the red mark on the chromosome), spores that are either homoallelic or heteroallelic for the gene can be produced, depending on the type of segregation. Specifically, if there is no recombination event between the gene and the centromere, the gene undergoes first division segregation (FDS) and the parental alleles co-segregate during meiosis I, generating homoallelic spores (i). FDS of a spore-killing gene will thus result in a two-spored ascus (ii). If there is a recombination event between the gene of interest and the centromere, second division segregation (SDS) occurs. In this case, heteroallelic spores will be formed (i). For spore-killing, a four-spored ascus will still be produced because only one copy of the spore killer is required to provide resistance (ii). As SDS is reliant on recombination, the frequency of SDS relates to the relative distance from the centromere and can be used for linkage mapping. When there is spore-killing, the percentage of two-spored asci is the frequency of FDS, and is referred to as the ‘spore-killing percentage’. The Psks were described by crossing different strains and evaluating their spore-killing percentage in each cross. The seven unique Psks were shown to interact in a complex hierarchy, showing either a dominance interaction, mutual resistance, or mutual killing. (See Appendix 1 for more details and Figure 4—figure supplement 1 for a reproduction of the hierarchy presented in van der Gaag et al., 2000).
Box 1—figure 1
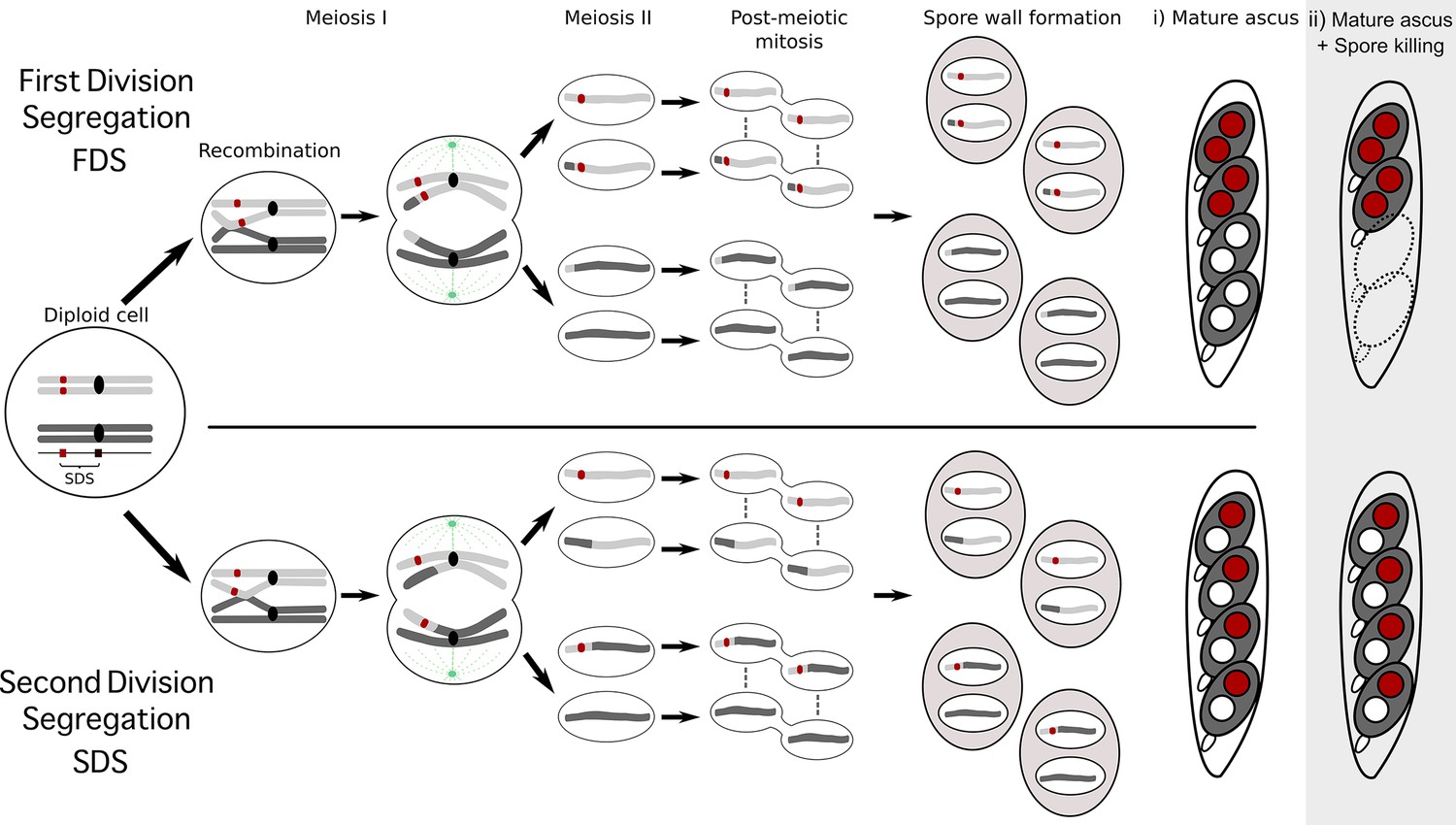
Schematic representation of meiotic segregation in Podospora anserina.
https://doi.org/10.7554/eLife.46454.003The primary goal of this study was to determine the identity of the genes that are responsible for the Psk spore-killer types found in P. anserina, and whether they relate to known meiotic drive genes. We identified two novel Spok homologs (Spok3 and Spok4) and showed that these two, together with the previously described Spok2, represent the genetic basis of the Psk spore killers. The new Spoks occur in large novel regions that can be found in different genomic locations in different strains. Our results illuminate the underlying genetics of a polymorphic meiotic drive system and expand our knowledge regarding their mechanism of action.
Results
Genome assemblies
To investigate the genetic basis of spore killing in P. anserina, we generated high-quality whole-genome assemblies using a combination of long-read (PacBio and MinION Oxford Nanopore) and short-read (Illumina HiSeq) technologies. Table 1 lists the strains used for investigation. In all cases, we sequenced single haploid monokaryons (marked with + or - following the strain name, to designate their mating type; see 'Materials and methods' and Appendix 1). We selected strains from a natural population in Wageningen (Wa), the Netherlands, and a few strains from France, representing six of the seven previously described Psk spore killer types (Psk-1, Psk-2, Psk-3, Psk-4, Psk-5 and Psk-7; van der Gaag et al., 2000) along with a strain of a novel killing type (Wa100), to which we assign the type Psk-8, and strain Wa63. The reference strain of P. anserina, S, was not given a Psk designation previously, as it was not known to induce spore-killing. However, Grognet et al. (2014) demonstrated that it can indeed induce spore-killing, so here we assign it to Psk-S along with Wa63. In addition, we acquired and sequenced strains from the closely related Podospora species P. pauciseta (CBS237.71) and P. comata (strain T). A strain annotated as T was acquired from two different laboratories, one from the Wageningen collection (referred hereafter as TD) and one from Goethe University Frankfurt (here as TG). Our results revealed that these strains do not represent the same isolate, as previously thought (Hamann and Osiewacz, 2004), but are distinct. TG is a Psk-5 strain of P. anserina and was sequenced with Nanopore and Illumina. TD matches the P. comata epitype reported by Silar et al. (2019) and was sequenced with Illumina alone.
Table 1
List of all strains used in this study.
https://doi.org/10.7554/eLife.46454.005| Sample | Site of origin | Spore killer* | Sequenced | Technology | Mycelium | Spok genes† | Spok block location‡ | Flanking genes§ |
|---|---|---|---|---|---|---|---|---|
| Natural isolates¶ | ||||||||
| Wa21– | Wageningen | Psk-2 (Psk-3) | DNA | PacBio | Monokaryon | Spok2, Spok3 | 5R: 3325285 | Pa_5_7950 – Pa_5_7960 |
| HiSeq 2500 | ||||||||
| Wa28– | Wageningen | Psk-2 | DNA | PacBio | Monokaryon | Spok2, Spok3 | 5R: 3325285 | Pa_5_7950 – Pa_5_7960 |
| HiSeq 2500 | ||||||||
| Wa46+ | Wageningen | Naïve (Psk-4) | DNA | PacBio | Monokaryon | SpokΨ1 | – | – |
| HiSeq 2500 | ||||||||
| Wa53– | Wageningen | Psk-1 | DNA | PacBio | Monokaryon | Spok2, Spok3, Spok4 | 3L: 358693 | Pa_3_945 – Pa_3_950 |
| HiSeq 2500 | ||||||||
| Wa58– | Wageningen | Psk-7 | DNA | PacBio | Monokaryon | Spok2, Spok3, Spok4 | 5L: 896822 | Pa_5_490 – Pa_5_470 |
| HiSeq 2500 | ||||||||
| Wa63+ | Wageningen | Psk-S | DNA | PacBio | Monokaryon | Spok2 | – | – |
| HiSeq 2500 | ||||||||
| Wa63– | Wageningen | Psk-S | RNA | HiSeq 2500 | Monokaryon | Spok2 | – | – |
| Wa87+ | Wageningen | Psk-1 | DNA | PacBio | Monokaryon | Spok2, Spok3, Spok4, SpokΨ1 | 3L: 358693 | Pa_3_945 – Pa_3_950 |
| HiSeq 2500 | ||||||||
| Y+ | France | Psk-5 | DNA | MinION | Monokaryon | Spok3, Spok4 | 3L: 358693 | Pa_3_945 – Pa_3_950 |
| HiSeq 2500 | ||||||||
| Wa100+ | Wageningen | Psk-8 | DNA | PacBio | Monokaryon | Spok2, Spok4, SpokΨ1 | 5L: 896822 | Pa_5_490 – Pa_5_470 |
| HiSeq 2500 | ||||||||
| TG+ | France | Psk-5 (sk-1) | DNA | MinION | Monokaryon | Spok3, Spok3, Spok4 | 3L: 358693 | Pa_3_945 – Pa_3_950 |
| DNA | HiSeq X | |||||||
| CBS237.71– | Israel | Psk-P1 | DNA | MinION | Monokaryon | Spok2, Spok3 | 4R: 1674812 | Pa_4_3420 – Pa_4_3410 |
| DNA | HiSeq X | |||||||
| TD+ | ? | Psk-C1 | DNA | HiSeq X | Monokaryon | Spok1 | – | – |
| RNA | HiSeq 2500 | |||||||
| S+ | France | Psk-S | DNA | HiSeq X | Monokaryon | Spok2 | – | – |
| S– | France | Psk-S | DNA | HiSeq X | Monokaryon | Spok2 | – | – |
| Wa47 | Wageningen | naïve (Psk-6) | – | – | – | Not sequenced | – | – |
| Z | France | Psk-7 | – | – | – | Not sequenced | – | – |
| s | France | Psk-S | – | – | – | Not sequenced | – | – |
| Us5 | Germany | Psk-S | – | – | – | Not sequenced | – | – |
| Backcrosses to S†† | ||||||||
| Psk1xS5- (Wa53) | Psk-1 | DNA | HiSeq 2500 | Monokaryon | Spok2, Spok3, Spok4 | 3L: 358693 | Pa_3_945 – Pa_3_950 | |
| Psk2xS5+ (Wa28) | Psk-2 | DNA | HiSeq 2500 | Monokaryon | Spok2, Spok3 | 5R: 3325285 | Pa_5_7950 – Pa_5_7960 | |
| Psk5xS5+ (Y) | Psk-1 (Psk-5) | DNA | HiSeq 2500 | Monokaryon | Spok2, Spok3, Spok4 | 3L: 358693 | Pa_3_945 – Pa_3_950 | |
| Psk7xS5+ (Wa58) | Psk-7 | DNA | HiSeq 2500 | Monokaryon | Spok2, Spok3, Spok4 | 5L: 896822 | Pa_5_490 – Pa_5_470 | |
| Psk1xS14-vsS | Psk-1 | RNA | HiSeq 2500 | Selfing dikaryon | Spok2, Spok3, Spok4 | Like parental | Like parental | |
| Psk2xS14-vsS | Psk-2 | RNA | HiSeq 2500 | Selfing dikaryon | Spok2, Spok3 | Like parental | Like parental | |
| Psk5xS14-vsS | Psk-1 | RNA | HiSeq 2500 | Selfing dikaryon | Spok2, Spok3, Spok4 | Like parental | Like parental | |
| Psk7xS14-vsS | Psk-7 | RNA | HiSeq 2500 | Selfing dikaryon | Spok2, Spok3,+ Spok4 | Like parental | Like parental |
-
*The spore killer type of each strain is given as reported by van der Gaag et al. (2000) when our phenotyping agrees, and in parenthesis when it does not. The S14 strains were phenotyped by us.
†The S14 Spoks were inferred from RNAseq mapping.
-
‡The chromosome number and the arm (R for right, and L for left) describing the position of the Spok block are given, along with the coordinates in the Podan2 chromosome.
§ As the exact insertion point is always intergenic, we also provide the flanking genes. The gene nomenclature follows Espagne et al. (2008), where Pa stands for Podospora anserina, the number between underscores is the chromosome and the last number is the gene code.
-
Note that strain s and strain S are different natural isolates.
††Parentheses denote parental spore killer strains.
-
‘Like parental’ denotes that the location of the Spok block in the S14 backcrosses was not inferred from sequencing data, however it should correspond to the location in the S5 backcrosses.
'–', Not applicable.
The final assemblies (long-read technologies polished with Illumina HiSeq data) recovered the expected seven chromosomes in their entirety for five strains, and in up to 13 scaffolds for the rest (Supplementary file 1). BUSCO analyses of these assemblies reported 97–98% of 3725 Sordariomyceta-conserved genes (Supplementary file 1), which is concordant with the same analysis done in the reference assemblies of the P. anserina strain S+ (hereafter referred to as Podan2; Espagne et al., 2008) and of P. comata (PODCO; Silar et al., 2019). Notice that as the assemblies of each strain were produced from one haploid (monokaryotic) isolate, we will refer to specific genome assemblies with their strain name followed by their corresponding mating type; for example, the assembled genome of monokaryon Wa63+ (derived from the strain Wa63) is called PaWa63p (Supplementary file 1).
In addition, all genomes sequenced with Illumina were assembled de novo using SPAdes. The resulting assemblies consisted of between 222 and 418 scaffolds that were larger than 500 bp, with a mean N50 of 227 kbp (Supplementary file 2). The alignment coverage of Podan2 (Espagne et al., 2008) was above 98% for all of the SPAdes assemblies of P. anserina. When the filtered Illumina reads were mapped to Podan2, all samples had a sequencing depth greater than 75x (Supplementary file 2). Taken together, our genome assemblies, resulting from both long- and short-read data, are very comprehensive.
The Podospora species are closely related and highly syntenic
A NeighborNet split network of 1000 single-copy orthologs (including introns) showed that the P. anserina samples are remarkably similar to each other and distinct from those of both P. comata and P. pauciseta (Figure 1A). Nevertheless, the three taxa are very closely related: the average genic identity within P. anserina is 99.97%, whereas the genic distance between P. anserina and P. pauciseta is 99.10%, between P. anserina and P. comata is 98.87%, and between P. pauciseta and P. comata is 98.79%. Accordingly, the whole-genome alignments recovered strongly conserved synteny at the chromosomal level when small (<13 kb) translocations (presumably due to transposable elements (TEs)) are excluded (Figure 1B; Figure 1—figure supplement 1). Only three larger translocations were detected, of which two might be mis-assemblies in the reference genome of P. comata (PODCO; see 'Materials and methods').
Figure 1 with 1 supplement see all
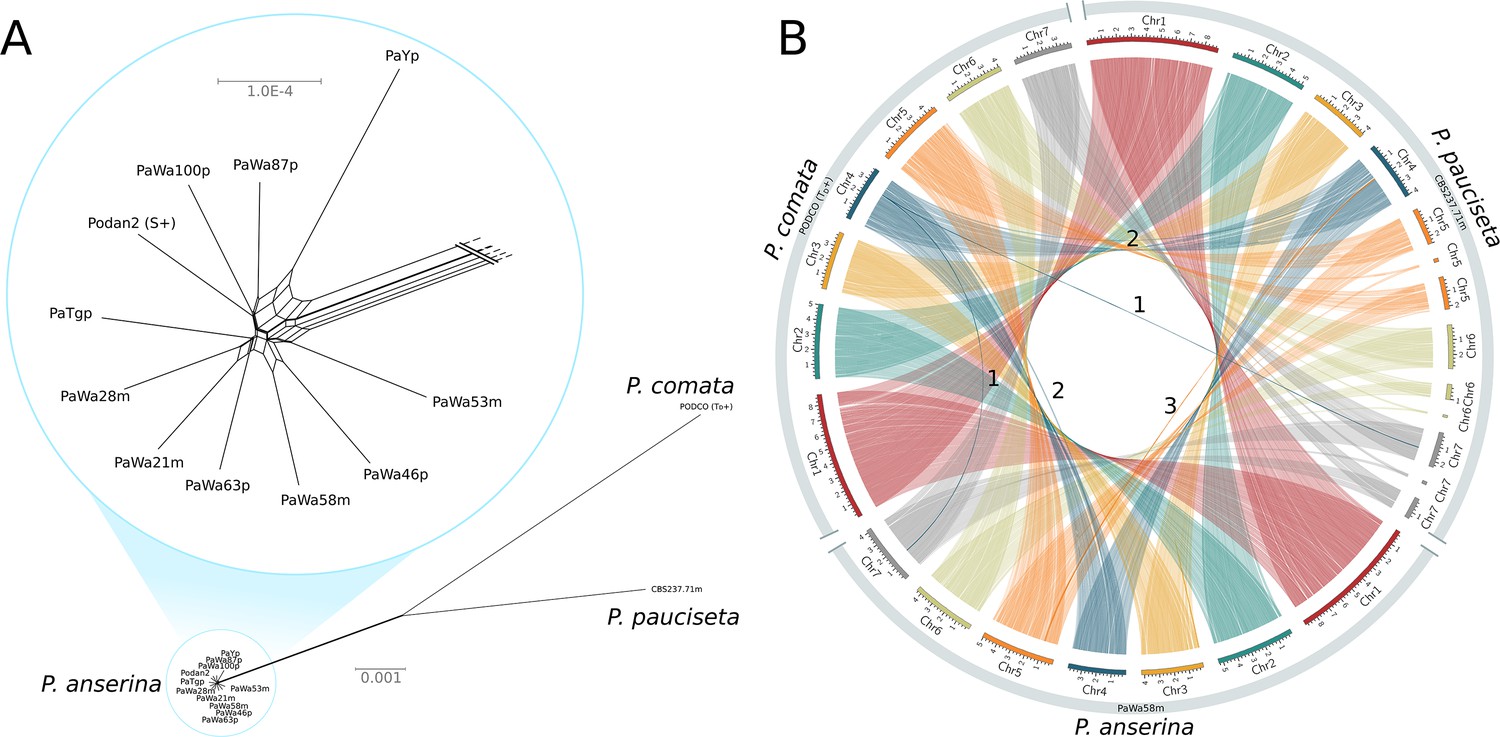
Relationship and synteny of Podospora strains.
(A) An unrooted NeighborNet split network based on 1000 orthologous genes of the strains representing the three Podospora species shows that the species are distinct from each other but still closely related. A close-up of the cluster of P. anserina strains reveals a reticulated relationship and very low genetic diversity (average genic distance of 99.97%). (B) A Circos plot of NUCmer alignments (larger than 13 kb) between the reference genome of P. comata, the new genome of P. pauciseta, and a representative strain of P. anserina. Chromosomes 5, 6, and 7 of P. pauciseta are not fully assembled, in particular around regions matching the location of the centromere in the P. anserina linkage map (not shown). Regardless, the alignment of the assembled region shows highly conserved large-scale synteny between the taxa, with the exception of three large translocation events marked with numbers. Numbers 1 and 2 are potential mis-assemblies in the P. comata reference genome, whereas the translocation number 3 between P. pauciseta and P. anserina corresponds to the Spok block. See Figure 1—figure supplement 1 for an equivalent Circos plot of inversions.
Identification and description of Spok genes
By searching our assemblies for the Spok2 sequence (presented by Grognet et al., 2014) using BLAST, we confirm the presence of this Spok gene on the left arm of chromosome 5 in the majority of strains, in agreement with Grognet et al. (2014). Furthermore, on the basis of sequence similarity with Spok2, we identified two novel homologs that we refer to as Spok3 and Spok4. These newly identified Spoks are found at different genomic locations depending on the strain. Both Spok3 and Spok4 can be located on the left arm of chromosome 3 or on the left arm of chromosome 5, and Spok3 can be found at an additional location on the right arm of chromosome 5. In addition, the BLAST searches recovered a pseudogenized Spok gene (SpokΨ1) in the subtelomeric region of the right arm of chromosome 5. The Spok gene content of the strains investigated in this study is reported in Table 1.
A schematic representation of the Spok homologs is shown in Figure 2A. We considered the Spok2 sequence of S+, and the Spok3 and Spok4 sequences of Wa87+ as reference alleles for each homolog. Overall they show a high degree of conservation, including the 3' and 5' UTRs. A nucleotide alignment of the Spok genes’ coding sequence revealed 130 variable sites out of 2334 total sites (Figure 2—figure supplement 1). A relatively large proportion (67%, 87/130) of those variable sites result in amino acid changes and 74% are unique to one of the Spok homologs. Table 2 displays pairwise comparisons of the amino-acid sequence of the SPOK proteins, revealing a high rate of non-synonymous substitutions, and a relatively high similarity between Spok1 and Spok4. There are six indels among all the Spok genes, including one at the 5' end of the ORF that represents a variable-length repeat region, and one at the 3' end of the ORF that is shared by Spok3 and Spok4. The 3' end indel induces a frameshift and changes the position of the stop codon (Figure 2A). SpokΨ1 has a missing 5' end, multiple stop codons, and a discoglosse (Tc1/mariner-like) DNA transposon (Espagne et al., 2008) inserted in the coding region. Of particular interest, SpokΨ1 has no deletions relative to the other Spok homologs, suggesting that the indels in the functional Spok homologs represent derived deletions.
Figure 2 with 2 supplements see all
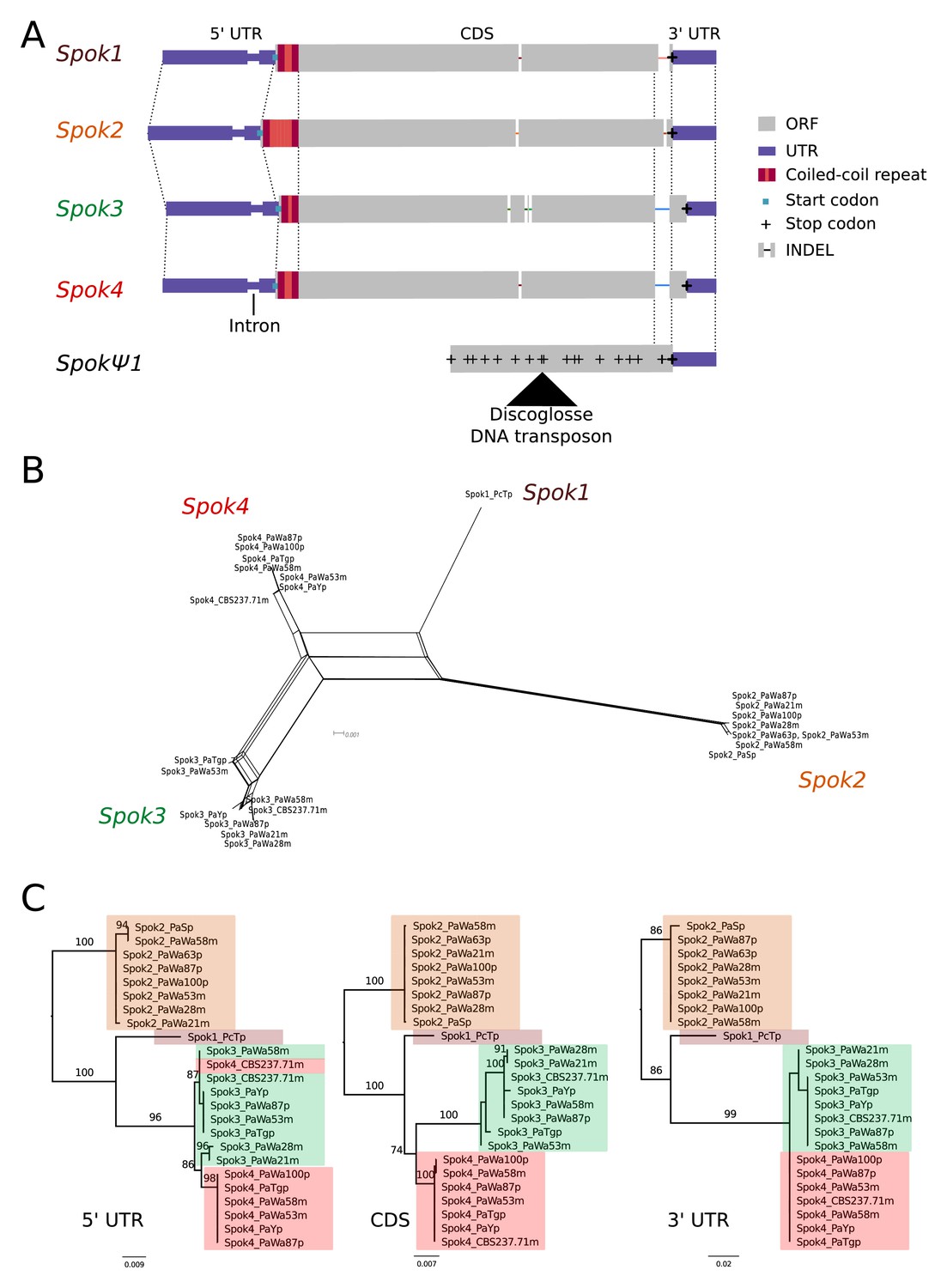
Relationships among the Spok homologs.
(A) Schematic representation of the main features of the Spok genes. All homologs share an intron within the 5' UTR. At the start of the coding region (CDS) there is a repeat region, in which the number of repeats varies among the homologs. The central portion of the CDS has a number of indels, which appear to be independent deletions in each of Spok2, Spok3, and Spok4. There is a frameshift mutation at the 3' end of the CDS that shifts the stop codon of Spok3 and Spok4 into what is the 3' UTR of Spok1 and Spok2. The pseudogenized Spok gene (SpokΨ1) contains none of the aforementioned central indels and appears to share the stop codon of Spok1 and Spok2. However, there are numerous mutations that result in stop codons within the CDS as well as a full DNA transposon (discoglosse) insertion. No homologous sequence of the 5' end of SpokΨ1 is present. (B) A NeighborNet split network of all active Spok genes from all strains sequenced in this study. The four homologs cluster together well, but there are a number of reticulations, which presumably are the result of gene conversion events. (C) Maximum likelihood trees based on three separate regions of the Spok genes: the 5' UTR, the CDS, and the 3' UTR (starting from the stop codon of Spok3 and Spok4). The trees are rooted arbitrarily using Spok2. Branches are drawn proportional to the scale bar (substitutions per site), with bootstrap support values higher than 70 shown above.
Table 2
Pairwise statistics between SPOK homologs.
The dN/dS ratios, averaged across the coding region, are shown below the diagonal; pairwise amino acid changes are shown above.
| SPOK4 | SPOK3 | SPOK2 | SPOK1 | |
|---|---|---|---|---|
| SPOK4 | x | 41 | 53 | 19 |
| SPOK3 | 0.8404081 | x | 54 | 51 |
| SPOK2 | 0.9731409 | 0.9771488 | x | 40 |
| SPOK1 | 0.6593501 | 0.7833958 | 0.7851462 | x |
There is little allelic variation within the Spok homologs in the Wageningen population and the variants of the four homologs cluster phylogenetically (Figure 2B and C). The Spok2 gene in the Wageningen strains are identical to the two alleles described in Grognet et al. (2014), with the exception of Spok2 from Wa58– which has a single SNP that results in a D358N substitution. The Spok2 allele of the French strain A, which shows resistance without killing (as reported by Grognet et al., 2014), was not found in any of the genomes investigated in this study. Spok3 has five allelic variants, and the allelic variation of Spok4 is reminiscent of Spok2, with only Wa100+ and Wa58– having a single synonymous SNP (Figure 2C). Lastly, the three copies of SpokΨ1 are all unique.
Notably, a number of the allelic variants of Spok3 show signatures of gene conversion events (Lazzaro and Clark, 2001). Specifically, strain Y+ has three SNPs near the start of the gene that result in amino-acid changes and that match those in Spok2 exactly (Figure 2—figure supplement 1). The Wa53+ allele of Spok3 has a series of SNPs (a track of 205 bp) that are identical to those in Spok4 but different from all other Spok3 sequences, and three additional SNPs near the 5' end that also match Spok4 (Figure 2—figure supplement 1). The TG+ strain possesses two identical copies of Spok3 (see 'Materials and methods') that share the aforementioned tract with Wa53+, but which extends for an additional 217 bp (Figure 2—figure supplement 1). These chimeric Spoks are recovered from the final assemblies (pre- and post-Pilon polishing) with high long-read coverage (>30 x), suggesting that our finding is not a bioinformatic artifact. The gene conversion events between Spok homologs are supported by the reticulation shown in a NeighborNet split network (Figure 2B) and by a significant recombination Phi test (199 informative sites, p=1.528e-12). A maximum likelihood phylogenetic analysis of the UTR sequences (defined by conservation across homologs) suggests that Spok3 and Spok4 are closely related (Figure 2C), which is at odds with the high structural similarity of the coding sequences of Spok1 and Spok4 (Figure 2A). Therefore, we cannot make any strong inference about the relationships between the Spok homologs from the sequence data.
In the few strains with no copy of Spok2, analysis of the region suggests that this is a result of a one-time deletion (Figure 3). The annotation in the original reference genomes of TD and S is erroneous because of mis-assemblies and/or incomplete exon prediction, which were both corrected in our study using our own Illumina data and annotation pipeline, and then validated by the RNAseq expression data for TD. First, the flanking gene P_5_20 (marked as (1) in Figure 3) in P. pauciseta (CBS237.71) and P. comata (TD) is considerably longer than the P. anserina ortholog, which is truncated by a discoglosse (Tc1/mariner-like) DNA transposon (2). In the strains without Spok2 (Wa46, Y, and TG), the discoglosse transposon itself is interrupted and the sequence continues on the 3' end of a fragmented crapaud (gypsy/Ty3) long terminal repeat (LTR) element, which can be found in full length downstream of Spok2 in the other strains. This configuration implies that the absence of Spok2 constitutes a deletion (3), rather than the ancestral state within P. anserina. An alternative scenario would require multiple additional insertions and deletions of TEs and Spok2.
Figure 3
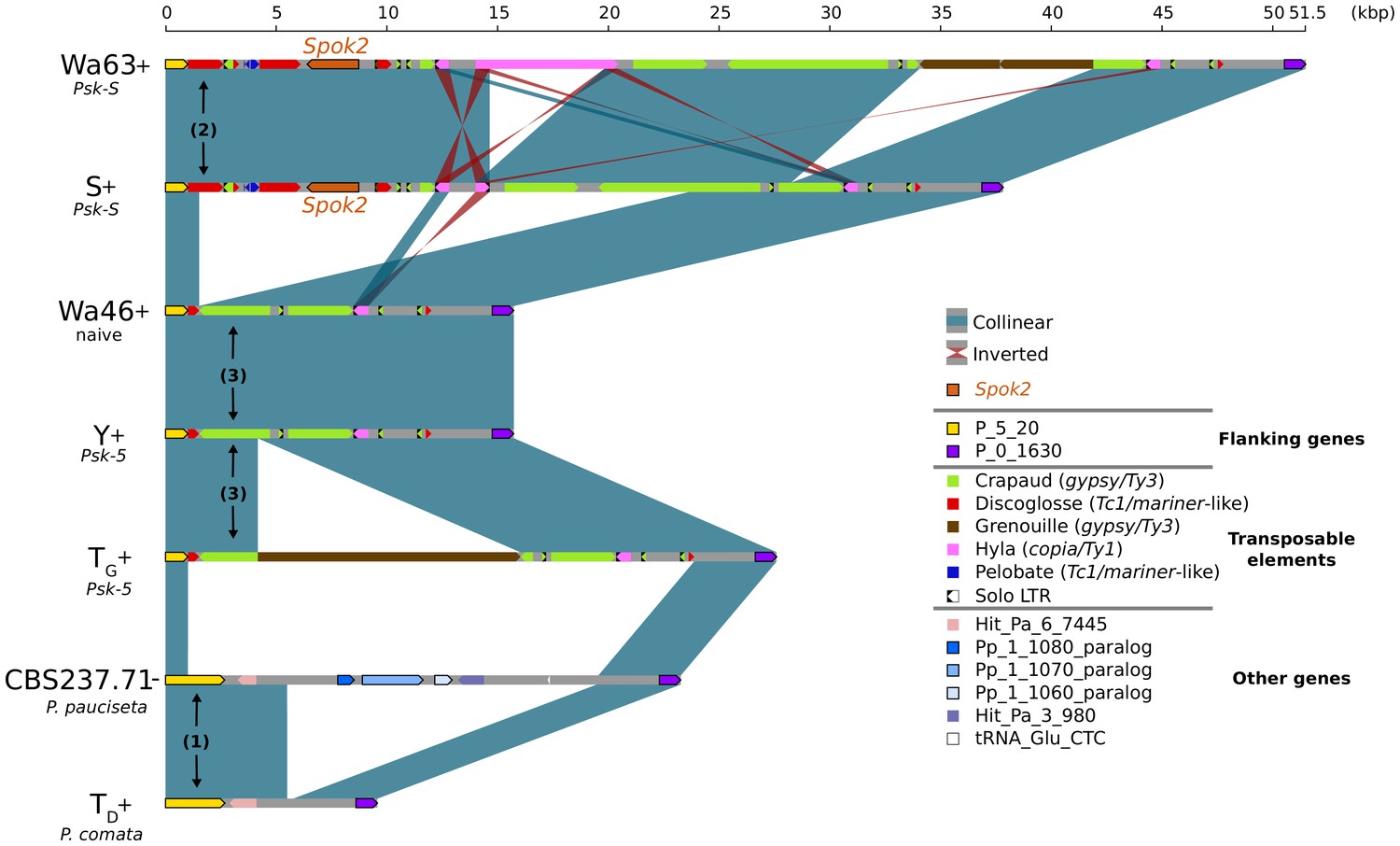
Alignment of the Spok2 locus in selected strains.
The plot displays pairwise comparisons of Spok2 haplotypes with blue segments connecting syntenic regions of homology and red segments showing inversions. The haplotypes are defined by the flanking genes P_5_20 and P_0_1630 located on chromosome 5 of the three sampled species. Every strain has a haplotype of different size, mainly owing to differences in their transposable element (TE) content. Within P. anserina, the TE variation across all sequenced strains occurs downstream of Spok2, as exemplified by strains Wa63 and S. The strains Wa46, Y and TG all lack Spok2 and share break points. See main text for a hypothesis of events (numbered). Notice that P_5_20 stands for the genes Pa_5_20 and PODCO_500020 in the reference annotation of P. anserina and P. comata, respectively, while P_0_1630 stands for Pa_0_1630 and PODCO_001630. As a note, P. pauciseta has a duplication of three genes in tandem from chromosome 1 (Pa_1_1080–60) between the flanking genes. Hit_Pa_X_XXX genes stand for significant BLAST hits to genes of Podan2. TE nomenclature follows Espagne et al. (2008).
-
Figure 3—source data 1
Annotation file for TEs surrounding Spok2.
- https://doi.org/10.7554/eLife.46454.013
The Spok1 gene was previously identified from the P. comata strain TD (Grognet et al., 2014). No other strains investigated in this study were found to possess Spok1, indicating that this gene is probably not present in P. anserina. Remarkably, BLAST searches of the Spok2 gene (including the UTR sequences) revealed the presence of a small piece (~156-bp long) of a presumably degraded Spok gene in the TD de novo assembly and on chromosome 4 of PODCO. This piece overlaps with the last amino acids of the CDS 3' end and is flanked by an arthroleptis (solo LTR) retrotransposon on one side and by unknown sequence on the other. Owing to the small size of this piece, it not clear whether it belongs to a novel Spok gene, but the location (between genes PODCO_401390 and PODCO_401400) differs from those of the other known homologs. The sequencing reveals that the genome of the P. pauciseta strain CBS237.71 contains both Spok3 and Spok4 (Table 1; Figure 2B), but they are at a genomic location that differs from those in any of the P. anserina strains.
Spok3 and Spok4 function as meiotic drive genes
We constructed knock-in and knock-out strains to confirm that the newly discovered Spok homologs, Spok3 and Spok4, can induce spore killing on their own (Supplementary file 3), as previously shown for Spok2 by Grognet et al. (2014). First, the Spok2 gene was deleted from the strain s to create a ΔSpok2 strain for use with the knock-ins. A cross between s and the ΔSpok2 strain resulted in about 40% two-spored asci, as previously reported by Grognet et al. (2014) (80/197, 40.6%) (Figure 4—figure supplement 2B). The Spok3 and Spok4 genes were inserted separately at the centromere-linked PaPKS1 locus (a gene controlling the pigmentation of spores [Coppin and Silar, 2007]). As PaPKS1 is tightly linked to the centromere, we expected that if the genes are capable of meiotic drive, then crosses to the ΔSpok2 strain should yield nearly 100% two-spored asci with white (unpigmented) spores. Accordingly, both Spok3::PaPKS1 ΔSpok2 x ΔSpok2 and Spok4::PaPKS1 ΔSpok2 x ΔSpok2 crosses yielded almost 100% two-spored asci with two white spores (118/119, 99.1%; Figure 4—figure supplement 2C) and (343/346, 99.1%; Figure 4—figure supplement 2D), respectively. These results show that Spok3 and Spok4 function as spore killers when introduced as a single copy at the PaPKS1 locus .
The P. anserina Spok homologs are functionally independent
To determine whether there are any interactive effects between Spok2, Spok3, and Spok4, we made use of the knock-in strains to assay pairwise interactions among them. First, to determine the interaction between Spok3 and Spok4, we crossed a strain bearing Spok4 at PaPKS1 with a strain bearing Spok3. Because crosses that are homoallelic for the PaPKS1 deletion have poor fertility, we constructed a strain in which Spok3 is inserted as a single copy at the PaPKS1 locus but just downstream of the coding region (Spok3::PaPKS1d) in order to yield strains with normal pigmentation and normal fertility in crosses to PaPKS1-deletion strains. In control crosses, the Spok3::PaPKS1d strain showed killing when crossed with a strain lacking Spok3 but no killing when crossed with Spok3::PaPKS1 (Figure 4—figure supplement 2E and F). The cross between Spok3::PaPKS1d and Spok4::PaPKS1 yielded asci that had four aborted spores, indicating mutual killing of Spok3 and Spok4 (Figure 4—figure supplement 2G). To determine the killing relation between Spok2 and Spok3, a cross was conducted between Spok3::PaPKS1 and strain s (of the Psk-S type). This cross yielded mostly two-spored asci with two unpigmented spores (163/165 asci: 98.8%) (Figure 4—figure supplement 2H), indicating that Spok3 kills in the presence of Spok2. Similarly, to determine the killing relation between Spok2 and Spok4, a cross was conducted between Spok4::PaPKS1 and s, which resulted in 99:5% killing (216/217 asci) (Figure 4—figure supplement 2I). Although these two crosses indicate that Spok2 does not confer resistance to Spok3 and Spok4 (Spok3 and Spok4 both kill Spok2), they do not allow us to determine whether Spok3 or Spok4 confer resistance to Spok2. To address this point, Spok2 killing was analyzed in a cross that was homoallelic for Spok3 (Spok3::PaPKS1 x Spok3::PaPKS1d ΔSpok2), which yielded 46% two-spored asci (143/310), confirming that Spok2 killing occurs in the presence of Spok3 (Figure 4—figure supplement 2J). Finally, to determine whether Spok4 is resistant to Spok2, we made a Spok4::PaPKS1 x Spok4::PaPKS1 ΔSpok2 cross, which resulted in 11/24 two-spored asci (Figure 4—figure supplement 2K). Although this genetic background is ill-suited for determining killing frequency (because of the aforementioned effect of the homozygous PaPKS1 deletion on fertility), the presence of two-spore asci suggests that Spok4 does not confer resistance to Spok2 killing. Overall, these results indicate that Spok2, Spok3, and Spok4 do not interact.
The Spoks are the Spore-Killer genes of the psks
To evaluate whether the newly discovered Spok homologs represent the genes that underlie the Psk spore-killer types, we sequenced backcrossed laboratory strains using Illumina Hiseq technology. A strain of each of Psk-1, Psk-2, Psk-5 and Psk-7 was previously backcrossed five times to the reference strain S (van der Gaag et al., 2000). The backcrossed strains are referred to here as Psk1xS5, Psk2xS5, Psk5xS5, and Psk7xS5 (Table 1). The backcrossed strains should maintain the killing percentage and mutual interactions of the dominant Psk parent. Given previous studies, we do not expect S (Psk-S) to be dominant over the other Psks (van der Gaag et al., 2000). Notably, crossing results reveal that Psk5xS5 has neither a Psk-5 nor a Psk-S phenotype, but a Psk-1 phenotype (Figure 4—source data 2). This is only possible if multiple killing loci are involved, which is concordant with the observation of multiple Spok genes in these strains.
Our Illumina data recovered a total of 41,482 filtered biallelic SNPs from the four S5 backcrosses and the parental strains. All backcrossed strains show a few continuous tracts of SNPs from the dominant killer parent (Figure 4—figure supplement 3). For example, Psk1xS5– has a long tract in chromosome 1 that represents the mat– mating type, which is to be expected because the published reference of S (Podan2), for which the SNPs are called, is of the opposite mating type (mat+). Importantly, the location of Spok3 and/or Spok4 of each parental strain has a corresponding introgressed SNP tract in the corresponding S5 backcross, while all backcrossed strains possess the Spok2 gene from strain S (Figure 4—figure supplement 3). The Psk-5 parental strain of Psk5xS5 (strain Y) does not possess Spok2, whereas Psk5xS5 does. Hence, the change in the killing phenotype of the backcrossed strain can be attributed to the presence of Spok2 (see below). Taken together, these data suggest that the total Spok gene content is responsible for the killer phenotype of Psk-1, Psk-2, Psk-5, and Psk-7 (Figure 4). In addition, we determined (on the basis of experimental crosses) that the newly described Psk-8 type can also be described by Spok gene content and position (Figure 4—source data 2 and 3). Specifically, Psk-8 has the same Spok block position as Psk-7, but does not possess Spok3 (Figure 4).
Figure 4 with 5 supplements see all
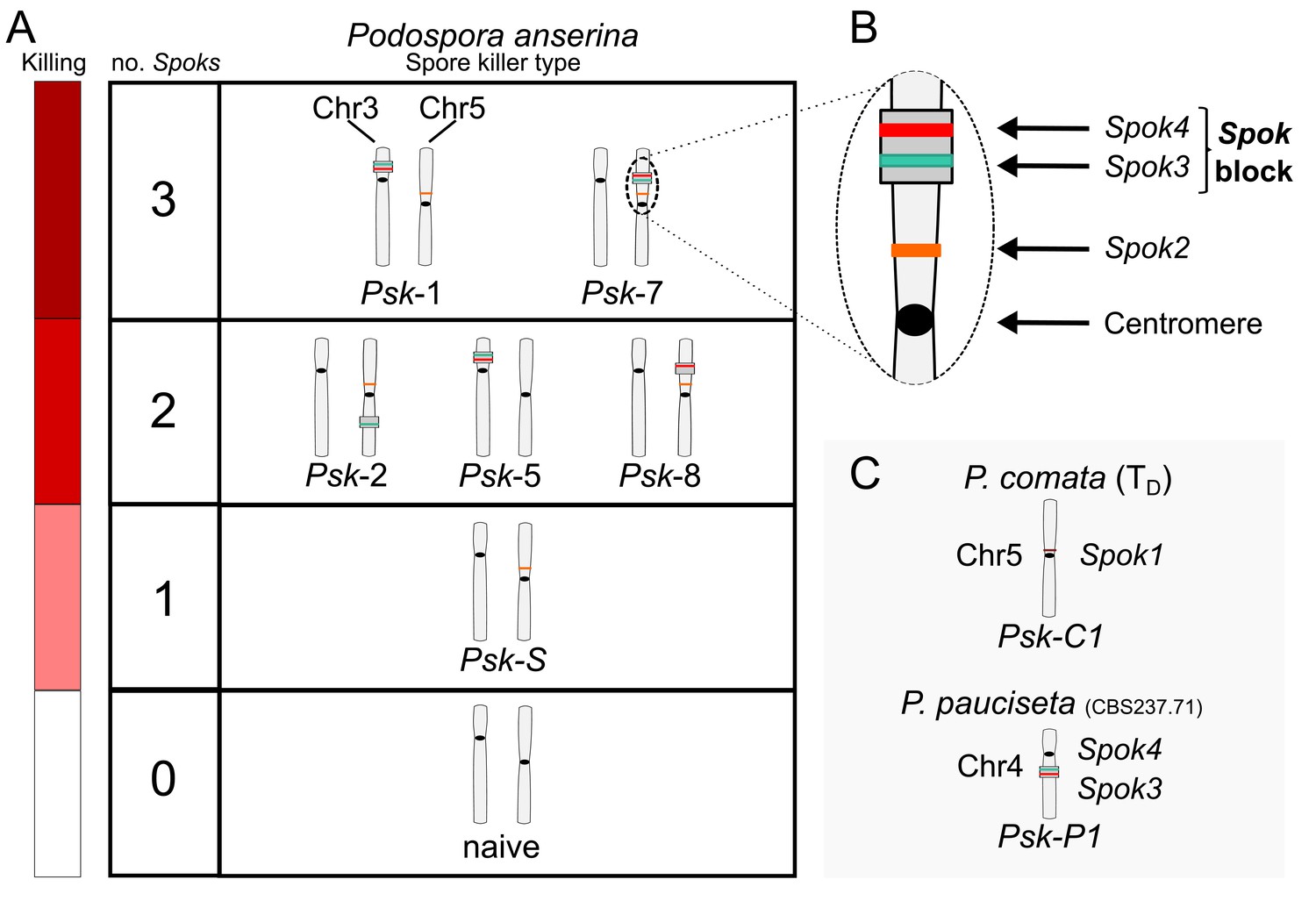
Interactions among the various Psk types and the occurrence of Spok genes.
(A) The boxes represent hierarchical levels that increase in killing dominance from bottom to top, which correlate with the number of Spok genes that a strain possesses. Strains with three Spok genes induce the spore-killing of strains with only two Spok genes and show mutual resistance to each other. Strains with two Spok genes show mutual killing among themselves due to the different Spok genes and kill strains with only Spok2. Strains with one Spok kill strains with no Spok genes (naïve strains). The chromosome diagrams depict the presence of the Spok genes and their location in the genome for the sequenced strains. (B) A zoomed-in look at Chromosome 5 of a Psk-7 strain, demonstrating that Spok3 and Spok4 are present in the Spok block and that Spok2 is present at the standard location. (C) The closely related species P. comata and P. pauciseta also possess Spok genes, but at different locations. The Spok genes in P. pauciseta are present in a smaller Spok block, whereas Spok1 is found on its own and exclusively in P. comata.
-
Figure 4—source data 1
Table showing which type of data was used to infer pairwise interactions amongst all Psks.
See Figure 4—source data 2 for killing percentages and Figure 4—source data 3 for backcross data.
- https://doi.org/10.7554/eLife.46454.020
-
Figure 4—source data 2
Table with killing percentages for all crosses tested between strains.
Values reported are FDS percentages with the total number of asci counted in parentheses.
- https://doi.org/10.7554/eLife.46454.021
-
Figure 4—source data 3
Table with observations of killing to determine pairwise interactions of the Psks.
Observations of spore killing or no spore killing are indicated for backcrosses to parental strains and from selfing. Empty cells indicate infertile crosses.
- https://doi.org/10.7554/eLife.46454.022
Our results from the crosses also identified inconsistencies with previous studies (see also Appendix 2). Originally, Psk-4 was defined as a spore-killer (van der Gaag et al., 2000. However, the Psk-4 strain Wa46 has no intact Spok genes (Table 1). The spore killing observed when this strain was crossed to S in previous publications (or to Wa63 here) is a result of Spok2-induced killing. Hence, we recommend discontinuing the use of Psk-4 and that the term ‘naïve’ strain is used instead. Moreover, our crossing data show that our representative strain of Psk-3 (Wa21) (van der Gaag et al., 2000) is of Psk-2 killer type because it does not exhibit spore killing when crossed to Wa28 (Psk-2), because it has the expected spore-killing percentage when crossed to a Psk-S strain, and because its Spok content and location are representative of a Psk-2 strain. Finally, our representative strain of Psk-6 (Wa47) behaves as naïve (Psk-4), and does not exhibit the spore-killing reported by van der Gaag et al. (2000) in test crosses with Wa46 (Figure 4—source data 2).
As each isolate of the entire Wageningen collection was previously assessed to determine its Psk type (van der Gaag et al., 2000), we can estimate the frequency of each Spok gene in the Wageningen population. Isolates of Psk-1, Psk-2, Psk-4, Psk-5, and Psk-7, as well as those previously considered as ‘sensitive’ (now Psk-S), account for 92 of the 99 strains collected from Wageningen. The seven remaining strains were identified as either Psk-3 or Psk-6. Following the rationale outlined in the previous paragraph, we assume that strains annotated as Psk-4 possess no functional Spok genes and omit all the Psk-3 strains (except Wa21) and the Psk-6 strains (except Wa47) from the analysis. We estimate that Spok2 is in 98%, Spok3 in 17%, and Spok4 in 11% of the Wageningen strains. A subsample of 11 strains from the 1937 French collection (including strains Y, Z and TG) have also been assessed for their Psk type , as have eight strains from a collection from Usingen (Us), Germany (Hamann and Osiewacz, 2004; van der Gaag et al., 2000). Hence, we infer that Spok2 is present in all of the Us strains and in 73% of the analyzed French strains. Spok3 and Spok4 is in 36% of the French strains, whereas Spok3 is in one Us strain and Spok4 is absent from the Us strains.
Spok3 and Spok4 are found in a large region associated with the Psk phenotypes: the Spok block
Although the Spok genes are often assembled into small fragmented contigs when obtained by using Illumina data alone, in the PacBio and MinION assemblies, Spok3 and Spok4 are fully recovered within an inserted block of novel sequence (74–167 kbp depending on the strain), hereafter referred to as the Spok block (Figure 4). When present, the Spok block was found once per genome and always contained at least one Spok gene. Whole-genome alignments revealed that the Spok block has clear boundaries, and is localized at different chromosomal positions on chromosome 3 or on either arm of chromosome 5 in different strains of P. anserina (Table 1). Importantly, these positions correspond to a single SNP tract identified in the S5 backcrosses. In P. pauciseta (CBS237.71), the Spok block is found on chromosome 4. This is evident in Figure 1B as the only large-scale translocation between P. anserina and P. pauciseta (number 3). The Spok blocks of the different strains share segments and overall structure (Figure 5 and Figure 5—figure supplement 2), which suggests that they have a shared ancestry. However, complex rearrangements are found when aligning the block between the genomes. Within the Spok block, a given strain can harbor either or both of Spok3 and Spok4, and the regions containing the Spok genes appear to represent a duplication event (Figure 5). Strain TG+ shows an additional duplication that has resulted in a second copy of Spok3 (Figure 5—figure supplement 2). When present, SpokΨ1 is surrounded by numerous TEs, and the region does not appear to be homologous to the Spok block (Figure 5—figure supplements 1 and 3).
Figure 5 with 3 supplements see all
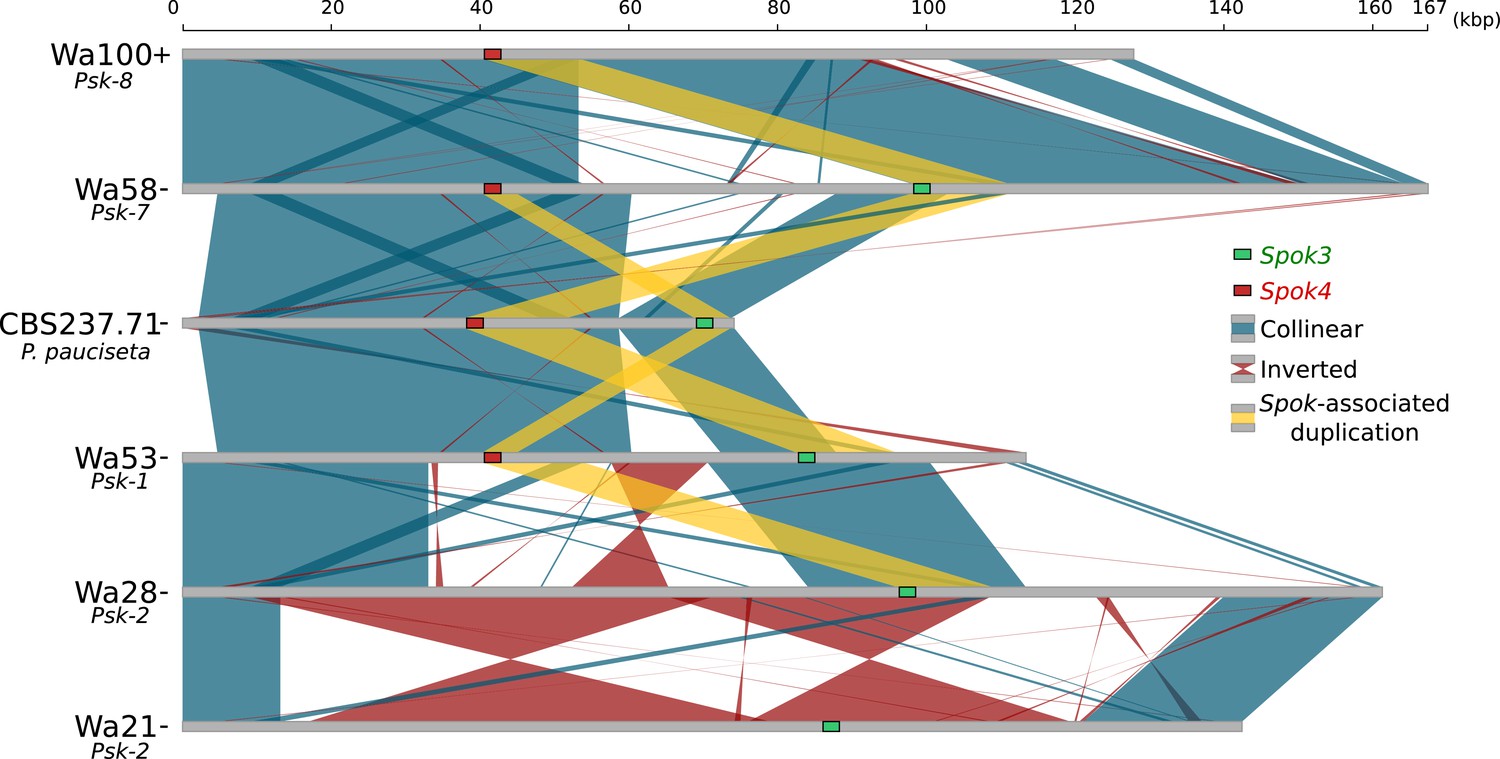
Alignment of the Spok blocks from different strains.
Gray bars represent the block sequences, blue vertical lines connect collinear regions between blocks, while red lines indicate inverted regions. The yellow lines show the region that is duplicated within the block surrounding Spok3 (green) and Spok4 (red).
All Psk interactions can be explained by the presence, absence, or location of Spok2, Spok3, and Spok4
To determine whether other components of the Spok block influence the interactions of the Spok genes and/or the Psks, we conducted crosses between selected strains and evaluated spore killing (Figure 4—source data 1, 2 and 3). Specifically, dikaryotic F1 progeny that are homoallelic for the killing locus were selected, backcrossed to both parental strains, and also allowed to self-fertilize (Figure 4—figure supplement 4). On the basis of the results of these crosses, killing interactions were classified into one of the following categories.
Mutual killing — spore-killing is observed when F1 strains are backcrossed to either parent, or when F1 progeny are selfed (Figure 4—figure supplement 4B).
Mutual resistance — no spore-killing is observed when F1 strains are backcrossed to either parent nor when selfed.
Dominance interaction — spore-killing is observed when F1 strains are backcrossed to only one of the parental strains, and no spore killing is observed upon selfing.
As an example, in a cross between strains of Psk-1 and of Psk-7 killer types, there is spore-killing. However the F1 progeny from this cross show no killing when crossed to either parent, nor when selfed, satisfying condition 2. Thus they are mutually resistant, which is consistent with the fact that they carry the same three Spok genes. The reason spore killing is observed in the original cross is because the Spok block is located at different genomic positions. As a result, the Spok block can co-segregate during meiosis, leaving two spores without any Spoks and making them vulnerable to killing (see Appendix 1 for a detailed explanation). An example in which the Psks show dominance is in the interaction between Psk-7 and Psk-8, which is revealed by the absence of killing when an F1 progeny from a Psk-7 x Psk-8 cross is selfed or crossed to the Psk-7 parent, and the observation of killing when crossed to the Psk-8 parent. This result indicates that the F1 progeny inherited its killing function from the Psk-7 parent, and is consistent with the idea that Spok gene interaction determine killing as, although Psk-8 and Psk-7 strains have a Spok block in the same location, the Psk-8 version of the block does not possess Spok3 (Figure 4).
The backcrossing method described above was not conducted for representative strains of all pairwise interactions among the Psks because of the unmanageable number of crosses that would be involved and the difficulty in mating some strains. In cases where dominance was strongly suspected (on the basis of Spok gene distribution, such as with crosses to Psk-S or naïve strains), the killing percentage was used as a proxy for dominance because in crosses involving dominance, the killing percentage should reflect that of the dominant Psk (Figure 4—source data 2 and 3). Figure 4 displays these results as a killing hierarchy, in which strains of the Psk type at the top of the hierarchy are dominant to strains of the Psk type lower in the hierarchy and either mutually resistant to (level 3) or mutual killers of (level 2) strains on the same level. The exception is Psk-5, which exhibits mutual killing with Psk-S. Our classifications, based on crosses, show that the killing hierarchy observed in the Wageningen population of P. anserina is an emergent property of the presence and absence of the various Spok homologs in the different genomes. Hence, our data demonstrate that other components of the Spok block do not affect spore-killing.
Of note, crosses between Psk-1 and Psk-5 strains have a lower killing percentage than would be expected on the basis of the FDS of Spok2 (~25% instead of ~40%). We confirmed that Spok2 is completely associated with two-spored asci in these crosses using a pooled sequencing approach (Figure 4—figure supplement 5), an observation that is in line with the backcrossing results. However, we noted a high prevalence of three-spored asci (which were excluded from the analyses). We also observed three-spored asci in crosses between Psk-S and naïve strains. Despite low germination rates, we have been able to isolate a spore from a three-spored ascus in a cross between Psk-S and a naïve strain that has no copy of Spok2 (Appendix 2). Therefore, the three-spored asci are probably due to incomplete penetrance of the killing factor and support the conclusion that the spore killing observed in these crosses is caused by the same gene, Spok2. This result is consistent with findings presented in the study by van der Gaag (2005) that provided independent evidence for incomplete penetrance of spore-killing between S and Wa46 (Psk-S and naïve). The lower killing percentage is probably the result of asci with FDS of Spok2, which contain three or four spores instead of two.
Spok interactions among the Podospora species
In contrast to the absence of epistatic interactions among the Spok genes of P. anserina, Spok1 of P. comata and Spok2 were shown previously to interact epistatically (Grognet et al., 2014). To determine whether Spok1 is also dominant to Spok3 and Spok4, crosses were conducted between strain TD and strains of P. anserina. Although TD shows low fertility with P. anserina (Boucher et al., 2017), we were successful in mating TD to a number of P. anserina strains of different Psk spore-killer types (Figure 4—source data 2 and 3). Often, only a few perithecia were produced with limited numbers of asci available to count, but despite this obstacle, the crosses clearly demonstrate that TD is dominant over Psk-S and Psk-2 strains, and is mutually resistant to a Psk-5 strain. These results imply that Spok1 provides resistance to all of the Spok homologs in P. anserina and is capable of killing in the presence of Spok2 and Spok3, but not Spok4. The mutual resistance with the Psk-5 strain also demonstrates that Spok4 provides resistance against Spok1. Additional crosses were also conducted with the P. pauciseta strain CBS237.71, which were consistent for the Spok3 and Spok4 interactions (Figure 4—source data 2 and 3). As both TD and CBS237.71 have unique spore-killing phenotypes, we assign them the types Psk-C1 and Psk-P1, respectively.
An intron in the 5′ UTR is not required for spore killing
To investigate whether the Spok genes are expressed during spore-killing, we conducted an additional nine backcrosses of the S5 strains to S, in order to generate S14 backcrossed strains (see 'Materials and methods'). We produced RNAseq data for self-killing S14 cultures and mapped the reads to the final assemblies of the dominant killer parental strains (Figure 2—figure supplement 2A). The expression of the Spok genes is evident in this data and supports the presence of an intron in the 5' UTR of the Spok homologs (Figure 2 and Figure 2—figure supplement 2B–E). Given its conservation across the Spok homologs and as the wtf spore-killer system in S. pombe was described as involving two alternate transcripts of the same gene (Nuckolls et al., 2017; Hu et al., 2017), the role of the intron in Spok3 spore-killing activity was investigated. The intron was deleted in a plasmid bearing the Spok3::PaPKS1 deletion cassette by site-directed mutagenesis, and the modified plasmid was used to transform the ΔKu70 ΔSpok2 strain. Three transformants bearing the Spok3 lacking the intron sequence (Spok3 Δi) were crossed to a ΔSpok2 strain. As in the control cross with wildtype (wt) Spok3, in which close to 100% killing was found, we observed that 109/109 of the asci contained two unpigmented spores (Figure 4—figure supplement 2L). Thus, Spok3 Δi displays wildtype killing activity. We conclude from this experiment that the unspliced form of Spok3 is not required for normal killing activity, and neither does the killing and resistance function rely on an alternatively spliced form of this intron.
Functional annotation of SPOK3 predicts three ordered domains
In order to gain insights into the molecular function of the SPOK proteins, domain identification was performed with HHPred and a HMM profile based on an alignment of 282 Spok3 homologs from various Ascomycota species. The SPOK3 protein was predicted to be composed of three folded domains (located at amino-acid positions ~ 40 to 170, 210 to 400, and 490 to 700 in the protein) separated by two unstructured domains (~170 to 210 and 400 to 490) as shown in Figure 6. No functional identification was recovered for domain 1, but a coiled-coil motif was found in the N-terminal 40 amino acids and predicted to form a parallel dimer, which corresponds to the variable length repeat of the nucleotide sequences (Figure 2A). Domain 2 showed homology to a class of phosphodiesterases of the PD-(D/E)XK superfamily (~214 to 325) with the catalytic residues forming the PD-(D/E)XK motif spanning positions 219 to 240 in the SPOK3 sequence (Steczkiewicz et al., 2012). The best hit in HHPred was to the HsdR subunit of a type-I restriction enzyme from Vibrio vulnificus (Uyen et al., 2009). The sequences align in the catalytic core region in the PD-(D/E)XK motif and also around a QxxxY motif (294 to 298 in SPOK3) that was found to be important for nucleic-acid binding and nuclease activity (Sisáková et al., 2008) (Figure 6—figure supplement 2).
Figure 6 with 2 supplements see all

Functional annotation of the SPOK protein.
(A) (Top) A predicted domain diagram of the SPOK protein displays the N-terminal coiled-coil region (in magenta), the N-terminal domain of unknown function (in lilac), the two unstructured regions (in pink), the PD-(D/E)XK nuclease domain (in green), the cysteine cluster region (in orange) and the kinase domain (in red) with coordinates based on the alignment of all SPOK homologs. The positions of key residues and conserved motifs are indicated with the same color code. The mutations labels that are marked in bold correspond to the SPOK3 coordinates, except for the mutations on the SPOK2 of strain A. (Bottom) A plot of the pairwise nucleotide distances between all alleles of a given Spok indicates which regions of the protein are conserved or divergent, and where the polymorphisms within a single Spok gene are located. The predicted unstructured regions generally show greater divergence. (B) HMM profile derived from an alignment of 282 SPOK3 homologs from Ascomycota showing conserved residues. The domains identified in (A) are shown with the same color code, and key motifs and residues are underlined. The profile was generated with Web logo v3. (C) Comparison of the HHM profiles in the catalytic loop and DFG-motif region in eukaryotic protein kinases and Kdo kinase (an ELK) with the same region in Spok homologs. The sequence below corresponds to the SPOK3 sequence.
-
Figure 6—source data 1
Transformation efficiency of Spok3 manipulations.
- https://doi.org/10.7554/eLife.46454.030
Domain 3 was identified as a hypothetical kinase domain (~539 to 700) as predicted previously by Grognet et al. (2014). In addition, a motif with a cluster of three highly conserved cysteine residues together with histidine residues (C-x3-C-x13-C-x5-H-x7-H) that is reminiscent of the zinc-finger motifs was identified upstream of the kinase motif (Figure 6). As previously reported for Spok2, D667 was identified as the catalytic base residue in the catalytic loop (subdomain VIb) of the kinase domain. Kinases often use other proteins as substrates, but they may also target small molecules (Smith and King, 1995). Inspection of the VIb and VII functional regions, which are informative as regards to kinase substrate specificity, suggests that the Spok-kinase domain might be more closely related to eukaryotic-like kinases (ELKs) than to eukaryotic protein kinases (ePKs), raising the possibility that this kinase domain is not necessarily a protein kinase domain and could phosphorylate other substrates (Steczkiewicz et al., 2012; Kannan et al., 2007).
The killing and resistance functions can be attributed to separate domains
The ability of the Spok genes to perform both killer and resistance functions with a single protein is unique among meiotic drive systems (Bravo Núñez et al., 2018b). To investigate the role that the domains 1–3 may play in these two functions, we constructed a number of point mutations and truncation variants of Spok3 and assayed their ability to kill or provide resistance in vegetative cells. We were able to determine that domain 2 is important for killing activity whereas domain 3 is important for resistance activity.
It had been shown previously that the predicted kinase domain of SPOK2 (Figure 6) is involved in the resistance function (Grognet et al., 2014). We introduced a point mutation in a plasmid-cloned Spok3 gene that led to the replacement of the predicted catalytic aspartic acid residue of Spok3 by an alanine (D667A). The mutant allele was first used to transform a ΔSpok2 recipient strain. This Spok3 D667A mutant allele leads to a drastic reduction in transformation efficiency (Figure 6—source data 1), whereas the Spok3 wildtypeallele only moderately affects the number of transformants. As this approach results in random integration and potential multicopy insertion, we also attempted to introduce the mutant Spok3 D667A allele as a single copy at the PaPKS1 locus, as described above for wildtype Spok3. The initial transformants were heterokaryotic and displayed sectors of abnormal growth that corresponded to unpigmented mycelium, presumably containing nuclei with Spok3 D667A that inserted at PaPKS1. Monokaryotic transformants could be recovered and were tested for killing activity in a cross to a ΔSpok2. Four-spored asci with two white and two black spores were observed, suggesting that the D667A mutation abolishes spore killing. However, when the integrated Spok3 allele was amplified by PCR and sequenced, it appeared that the allele presents a GAG to TAG mutation, leading to a premature stop codon in position 282 (E282stop). This result is consistent with the observation that Spok3 D667A affects transformation efficiency and is toxic. Moreover, we detected expression of Spok2 and Spok1 in the vegetative cells of monokaryotic (self-sterile) cultures, suggesting that Spok activity is not restricted to the sexual cycle (Figure 2—figure supplement 2C and D). No further attempts to insert the mutant allele at PaPKS1 were made.
If the toxicity of the Spok3 D667A allele in vegetative cells is mechanistically related to spore-killing, it is expected that this toxicity should be suppressed by wildtype Spok3. Therefore, we assessed whether Spok3 D667A toxicity in vegetative cells is suppressed by co-expression with wildtype Spok3. Co-transformation experiments were set up with Spok3 D667A used as the transformation vector in the presence or absence of wt Spok3. As in the previous experiment, Spok3 D667A alone was found to affect transformation efficiency, but this effect was suppressed in co-transformations with Spok3 (Figure 6—source data 1). This experiment confirms that Spok3 D667A is only toxic in the absence of Spok3. Therefore, the Spok-related killing and resistance activities can be recapitulated in vegetative cells.
We also analyzed the role of the conserved cysteine cluster just upstream of the predicted kinase domain. Three plasmids with point mutations in that region were constructed (a C493A C497A double mutant, and C511A and C511S point mutants), and the mutant alleles were used in transformation assays as previously described for Spok3 D667A. All three mutants reduced transformation efficiencies as compared to the controls, and this effect was suppressed in co-transformations with wt Spok3 (Figure 6—source data 1). These results suggest that the kinase domain and the cysteine-cluster region are both required for the Spok-related resistance function but not for the killing activity. To test this, we constructed a truncated allele of Spok3 that lacks these two regions: Spok3(1–490) (see Figure 6—figure supplement 1). The Spok3(1–490) allele drastically reduced transformation efficiencies and this effect was suppressed in co-transformations with wildtype Spok3 (Figure 6—source data 1). If, as proposed here, the toxicity and suppression activities assayed in vegetative cells are mechanistically related to spore-killing, then domain 3 appears to be required for the resistance function but dispensable for the killing activity, which can be carried out by the N-terminal region of the SPOK3 protein (domains 1 and 2).
Next we analyzed the role of the predicted nuclease domain (domain 2) in spore-killing activity. We generated a plasmid with a point mutant that affects the predicted catalytic core lysine residue (K240A). Introduction of this point mutation in the Spok3(1--490) allele abolished its killing activity in transformation assays (Figure 6—source data 1), suggesting that the predicted nuclease domain is required for killing activity. The Spok3 K240A mutant was then inserted at the PaPKS1 locus and the resulting knock-in strain was crossed with a ΔSpok2 strain (in order to assay killing) and to a Spok3::PaPKS1d strain (to assay resistance) (Figure 4—figure supplement 2M and N). In the cross to ΔSpok2, no killing was observed: the majority of the asci were four-spored with two white and two black spores (308/379, 81.2%), indicating that the K240A mutation abolishes the spore-killing activity of Spok3. In the Spok3 K240D::PaPKS1 x Spok3::PaPKS1d cross, no killing was observed: the majority of the asci were four-spored with two white and two black spores (268/308, 87%). These crosses indicate that the Spok3 K240A allele has no killing ability but has retained resistance. Grognet et al. (2014) reported that strain A bears a mutant allele of Spok2 that has affected killing ability but retains resistance. The mutations in that allele fall within a conserved region of the predicted nuclease domain (Figure 6) and map on predicted structural models in close vicinity to the catalytic lysine residue (K240 in SPOK3) and the other catalytic residues (Figure 6—figure supplement 2). Hence, the properties of the Spok2 allele of strain A provide independent evidence that the nuclease domain of SPOK proteins is involved in killing activity but dispensable for resistance.
Phylogenetic distribution of Spok genes
A BLAST search for closely related homologs of the Spok genes across fungi revealed an uneven distribution of related proteins among taxa. Among the available genomes from the Sordariales, we did not find any protein-coding sequences in addition to our newly described Spok genes of Podospora. By contrast, we found protein-coding sequences with high similarity across other orders of the Sordariomycetes, namely the Xylariales, Glomerellales and Hypocreales, as well as in one species of the Eurotiomycetes, Polytolypa hystricis (Poh; Figure 7).
Figure 7
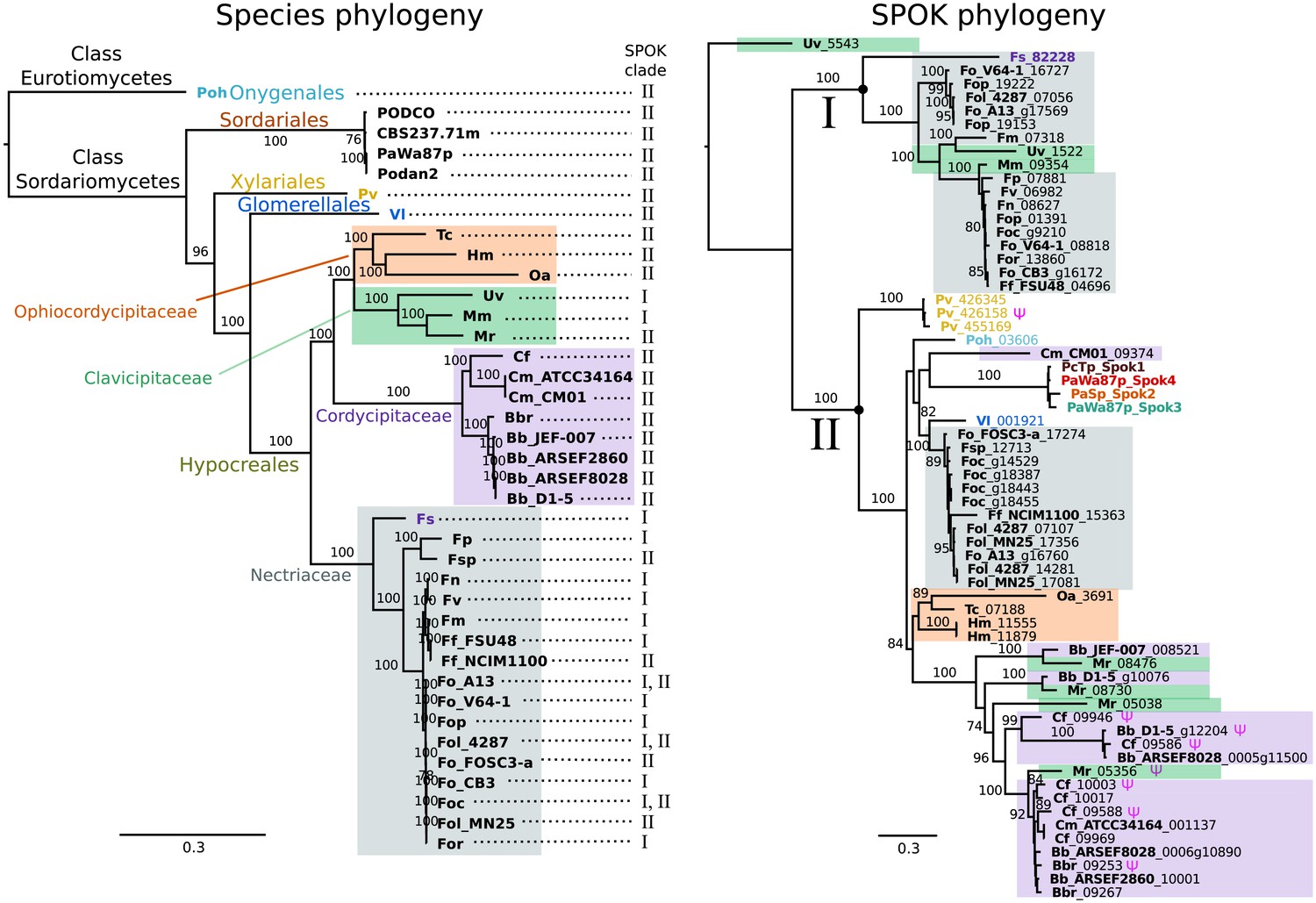
The phylogeny of Podospora SPOKs and closely related homologs do not follow the species tree.
(Left) A maximum likelihood phylogeny of the fungal isolates that harbor Spok homologs that are closely related to those of Podospora recovered the groupings that are expected on the basis of fungal taxonomic classification (colored boxes and tip labels). The tree was produced using the aligned protein sequence of 288 single-copy orthologs. (Right) A maximum likelihood phylogeny of the SPOK proteins themselves with colors matching the taxonomy in the tree to the left. Two main clades can be distinguished (I and II), and their presence is mapped to each genome in the species phylogeny. Putative pseudogenes are marked with a Ψ symbol. The Fs_82228 protein (in dark purple text) has been demonstrated to exhibit some spore-killing characteristics in a P. anserina strain. Rooting of the species tree was based on the split between Classes, whereas the SPOK phylogeny was rooted on the basis of the broader alignment generated for the protein-domain predictions. Bootstrap support values higher than 70 are shown above branches, which are proportional to the scale bar (substitutions per site). SPOK tip labels follow the convention of fungal isolate code (bold) and locus name (see Figure 7—source data 1 for full species names and genomes).
-
Figure 7—source data 1
Table showing species names and genome labels for codes used in Figure 7.
- https://doi.org/10.7554/eLife.46454.032
We used maximum likelihood analyses to construct phylogenies of the SPOK sequences and of an orthologous gene set of the strains for which we retrieved hits in the BLAST search (Figure 7). These phylogenies reveal two notable patterns. First, the SPOK phylogeny shows a high degree of incongruence with the species phylogeny. Moreover, the SPOK phylogeny can be robustly divided into two clades: Clade I and II. Clade I contains the Fs_82228 sequence from Fusarium solani (old name Nectria haematococca). This sequence was previously introduced into P. anserina, and the genetically modified strain produced empty asci when mated to a naïve strain, suggesting that it has a killing action (Grognet et al., 2014). Clade II contains the Podospora Spok homologs. The sequences from the two clades are disparately distributed in the species phylogeny. The second notable pattern is the distribution of the SPOK sequences within the genomes. The sequences in Clade I are present in single copies in each strain, except for Fusarium oxysporum f. sp. pisi (Fop), suggesting that they are all orthologs. By contrast, many of the sequences in Clade II are present in multiple copies in each genome. It is particularly interesting to note how many Spok homologs from Clade II are present across strains of F. oxysporum (Fo) and the number of copies that are found in each genome. Several of the duplicate Spok homologs are present on the lineage-specific chromosomes of Fusarium that are often associated with pathogenicity (Armitage et al., 2018). Beauveria bassiana (Bb) also shows a high degree of variability in homolog content among the four strains that have homologs, indicating that the homologs are polymorphic in this species. The insect pathogen Metarhizium rileyi (Mr) shows an interesting pattern in that it possesses four divergent homologs, which is in stark contrast to many of the other species(including Podospora) that have multiple, though nearly identical, copies. The Clade II Spok homologs also appear to diversify within each strain or species in much the same way as the Spok genes do in Podospora, with variable lengths of the coil-coil repeat region and frameshift mutations near the 3′ end that relocate the stop codon. A few of the sequences may also represent pseudogenes, as evidenced by premature stop codons and/or frameshifts, although these features might also be the result of unidentified introns (Figure 7).
Discussion
The identification of Spok3 and Spok4 has allowed us to explain the genomic basis for five of the seven Psk spore-killer types found in natural populations of P. anserina. Through our integrative approach of genomics, molecular biology and phenotyping, we have been able to demonstrate that the multiple drive elements that have been genetically identified in P. anserina are not based on different underlying molecular mechanisms and/or specific gene interactions, but rather involve combinations of closely related driver genes that belong to the same Spok gene family. The Spok genes thus appear to be responsible for all of the identified drive elements in Podospora, with the exception of the het-s spore-killing system.
The Spok block
The presence of the complex Spok block presents a unique feature among the known meiotic drive systems. Often, meiotic drive elements occupy regions of suppressed recombination that span large tracts of chromosomes (Turner and Perkins, 1979; Hammer et al., 1989; Sandler et al., 1959) and co-occur with complex rearrangements (Harvey et al., 2014; Silver, 1993; Dyer et al., 2007; Svedberg et al., 2018). In these well-studied cases, the elements of the drive mechanisms are encoded by separate genes within the region, and the rearrangements and suppression of recombination are expected to have evolved to ensure that the drive machinery (e.g., the toxin and antitoxin genes) is inherited as one unit (Lyttle, 1991; Bravo Núñez et al., 2018b). In Podospora, a single Spok gene is fully capable of driving, and thus no region of suppressed recombination is required. Nevertheless, Spok3 and Spok4 are found in a large region that is not syntenic with the null allele. Hence, had the Spok genes not been previously identified from more placid genomic regions, the entire Spok block may have been misidentified as a driving haplotype with multiple interacting components. Considering that single-gene meiotic drivers might be more common than anticipated, it becomes necessary to question whether other drive systems that are located within complex regions, and for which the genetics are not well known, may also represent single gene drivers.
At this stage, our data strongly suggest that the Spok block is moving in the genomes as a unit, but nevertheless, the mechanism of movement remains unknown. It may be hypothesized that movement of the block is achieved via an interaction with TEs at different genomic locations and non-allelic homologous recombination. This hypothesis is supported by the observation that the Spok genes outside of the Spok block, including SpokΨ1, are not located at the same position in different species, and that they are often surrounded by similar TEs. Such movement may be under selection as matings between strains that have the same Spok genes but in different locations will result in spore-killing. Furthermore, because of the idiosyncrasies of meiosis in Podospora, the position of the block may be under selection because the killing frequency is dependent on the frequency of crossing over with the centromere. Alternatively, the TEs may simply accumulate around the Spok genes because of a reduced efficacy of purifying selection at regions linked to the driver genes; their presence per se might increase the chance of rearrangements. As such, the role that TEs play in generating complex regions that are associated with meiotic drive should be investigated further in order to determine their importance in the evolution of drive.
Molecular function of the Spok genes
Spore-killing systems display analogies to toxin-antitoxin (TA) systems in bacteria and it is interesting to note that many toxin families rely on nuclease activity (Harms et al., 2018). The contrast between the Spok system and TA systems, however, resides in the fact that Spok toxin and antitoxin activities appear to be supported by the same protein molecule. The predicted kinase activity seems able to counter the toxic activity of the predicted nuclease domain both in cis and in trans. Mechanistic spore-killing models have to explain: (i) how the Spok gene can affect spores that do not carry it, at a distance, with SPOK proteins being intracellular proteins (Grognet et al., 2014); and (ii) how asymmetry is brought about in this system if killing and resistance activities are carried by the same protein molecule. It is premature to devise a mechanistic model for the molecular basis of Spok gene drive, yet it might be possible to conjecture about the substrates of the proposed kinase (and nuclease) activities of SPOK proteins.
A first way to explain how Spok genes might act at distance and affect spores not containing them is to hypothesize that toxicity relies on the production of a diffusible metabolite. The predicted nuclease/phosphodiesterase activity would lead to the production of a diffusible toxic molecule that could be further detoxified by phosphorylation (much in the way that bacterial phosphotransferases detoxify antibiotics) (Shi et al., 2013). Isolation of the toxic intermediate from the detoxifying activity of the kinase would allow its accumulation specifically outside of the spores expressing SPOK proteins, and thus could bring about asymmetry in spore-killing. Then, alternatively, spores lacking the Spok gene might be affected by SPOK proteins if some amount of SPOK protein expressed in the zygote or from sister nuclei is carried over at the time of spore delimitation. The resistance function of the predicted kinase domain could be explained by hypothesizing that the nuclease activity can be inhibited by autophosphorylation of the SPOK proteins. Alternatively, it could also be that it is the phosphorylation of a distinct macromolecule that nullifies toxicity. In a simple model, the same molecule could be the target of both the kinase and nuclease activity, and the phosphorylation of the target would make it resistant to the toxic action of the predicted nuclease domain. As stated above, models have to explain how killing could occur specifically in spores lacking the Spok gene. This situation could occur if the proposed kinase activity is concentration-dependent and favored at higher SPOK protein concentrations (for instance, the kinase activity might require a protein dimerization step that occurs specifically in spores expressing the Spok gene). In addition to the yet unresolved mechanistic basis of killing and resistance, the characterization of Spok gene function described here poses another puzzle. As all SPOK products have a predicted active kinase, it is not yet known what changes in sequence confer the hierarchical interactions among some Spok genes, or why not all SPOKs are able to provide resistance to one another. One possibility is that the cellular targets for the proposed nuclease and kinase activities differ for the different SPOK proteins.
Studies of similar protein domains suggest that the coil-coiled domain is likely to be involved in protein–protein interactions (van Maldegem et al., 2015). The fact that Spok1 and Spok4 have the same length repeat in this domain could imply that the protein–protein interactions of this domain are important for resistance, as Spok1 and Spok4 are mutually resistant. This model would agree somewhat with the results of reporter constructs from Grognet et al. (2014), which showed an N-terminal mCherry tag on Spok2-produced empty asci. It is possible that the functional divergence observed between the SPOK proteins is due to mutations in this portion of the protein. In this model, domain 1 might be responsible for the target specificity of the nuclease (and kinase) activity. The killing action itself is expected to be universal among the Spoks and is supported by the fact that this entire domain of Spok3 from TG is identical to Spok4, yet appears to retain Spok3 functionality. The identification of the role of the predicted nuclease domain in killing and of the predicted kinase domain in resistance provides a first mechanistic insight into the dual role of Spok genes. However, further dissection of the molecular action of these proteins is required so that we can fully understand the molecular basis of Spok drive.
Absence of resistance
One of the main factors that stands out in the Podospora system, as compared to the other well-studied spore killers, is the lack of resistant strains. Only one strain of P. anserina (the French strain A) has ever been described as resistant (Grognet et al., 2014). The point mutations of Spok3 that were induced in the laboratory imply that the creation of a resistant strain is a simple task, as only a single nucleotide change was required. Likewise, the resistant strain A Spok2 is different from the reference allele by only two novel insertions. Consequently, the lack of resistance does not appear to be the result of a mechanistic constraint. Potentially, the current Spok gene distribution could be a relatively young phenomenon and resistance could evolve over time. Another possibility is that resistance itself is somehow costly to the organism and selected against. In addition, it is puzzling that none of the Spoks in P. anserina show cross resistance. Intuitively, it would seem advantageous for novel Spok homologs to evolve new killing functions while maintaining resistance to the other Spok homologs. Again, the lack of cross-resistance does not solely appear to be the result of functional constraints, as Spok1, which is highly similar to Spok4, is resistant to all other Spok homologs. It is possible that it is more advantageous to combine multiple independent spore killers than to have a single broadly resistant gene. This option is supported by two observations presented in this study: the occurrence of the killing hierarchy and the association of Spok3 and Spok4. The fact that Spok3 and Spok4 are present in the Spok block means that they are in tight linkage with each other. It may be the case that the linkage was selected for because it provided strains with the ability to drive against strains with just Spok3 or just Spok4. However, this association could also be simply the result of a duplication without invoking selection. Whether the killing hierarchy that we observe in P. anserina is due to a complex battle among the Spok homologs or a result of the existence of the Spok block will require further experimentation and mathematical modeling to resolve.
Evolutionary dynamics of the Spok genes
Some interesting aspects of meiotic drive in Podospora identified herein bear numerous features that parallel the wtf genes that are responsible for drive in Schizosaccharomyces pombe. There is no sequence similarity or conserved domains between the Spok and wtf genes, and Podospora and Schizosaccharomyces are only distantly related (~500 million years diverged) (Wang et al., 2009; Prieto and Wedin, 2013). Yet these systems display similar evolutionary dynamics within their respective species. Both of these systems are built of multiple members of gene families, which appear to duplicate, rapidly diverge to the point where they no longer show cross reactions (potentially with the aid of gene conversion), and then pseudogenize and become nonfunctional (Nuckolls et al., 2017; Bravo Núñez et al., 2018a; Hu et al., 2017). Both systems also have close associations with TEs (Bowen et al., 2003). Hu et al. (2017) invoke LTR-mediated non-allelic homologous recombination as a possible mechanism for wtf gene deletion in a lab strain of S. pombe. We provide evidence for the deletion of Spok2, but it does not fit with expectation that this deletion is LTR-mediated. Nevertheless, as TEs are still accumulating in the region, other TE-related processes may have been involved in the deletion.
The factors that determine the abundance and diversity of multigene family meiotic drivers in a species are the rates of gene duplication and loss, and time since origin. In the case of the Spok genes, we expect a low rate of deletion as they approach fixation because of the dikaryotic nature of Podospora. Specifically, when first appearing, a deletion is only expected to be present in one of the two separate nuclear genomes maintained within a dikaryon. Any selfing event should erase (i.e. drive against) the deletion, meaning that in order to become homoallelic for a deletion, the strain would have to outcross with another individual with no Spok genes or Spok genes that differ from its own. Such outcrossing could allow deletions of Spok3 and Spok4, but as Spok2 is nearly fixed in the population, any outcrossing event should also lead to the elimination of the deletion by the driving action of Spok2. A possible solution to the paradoxical finding that Spok2 appears to have been lost occasionally is that the incomplete penetrance of Spok2 may have allowed spores that were homoallelic for the deletion to survive and persist. In this sense, Spok2 fits a model of driver turn over, wherein it is beginning to lose killing function after becoming fixed in the population. SpokΨ1 is missing the portion of the gene that is responsible for killing and the small Spok fragment of P. comata also corresponds to the resistance part of the gene. Both of these observations suggest that the killing domain may have been lost prior to these genes becoming fully pseudogenized and hints that they may have functioned as resistance genes.
It has been pointed out that spore-killing may be a weak form of meiotic drive, because the transmission advantage is relative to the number of spores produced in a given cross, but there is no absolute increase at the population level (Lyttle, 1991). Hence, a spore killer was predicted by Nauta and Hoekstra (1993) to require an additional fitness advantage in order to reach fixation in a population. It is thus striking that Spok2 is close to fixation in at least the European populations, bringing into question the direct fitness effects of the Spok genes. On the other hand, the Spok block (and hence Spok3 and Spok4) seems to be present at relatively low frequency. It is possible that the rate at which the Spok block switches position is higher than the rate at which the Spoks can sweep to fixation. Therefore, the dynamics of Spok genes within the Spok block might differ from the Spok2 life-cycle and might explain why spore-killing is observed to be polymorphic in P. anserina. In addition, P. anserina is capable of selfing, which may slow down the rate of fixation of the genes. Moreover, the vegetative and/or sexual expression of the Spok genes might be deleterious in itself, and hence natural selection might increase or maintain the frequency of strains without all Spok homologs. Overall, this complex system requires population genetic modeling to resolve the factors affecting the frequency of the Spok genes in populations of this fungus.
The relationships among the Spok genes can provide insight as to the evolutionary history of the Spok block. The observation that Spok3 and Spok4 are both present in the Spok block in a duplicate region suggests that these genes represent paralogs that formed via duplication. Indeed, the phylogenetic analysis of the UTRs agrees with a duplication origin. However, this scenario is contradicted by the finding that Spok4 shares many features with Spok1 of P. comata, but not Spok3. The four most likely evolutionary scenarios are outlined in Figure 8. If the relationship between Spok1 and Spok4 is a result of common descent (orthology; Figure 8A), then after the duplication event that generated Spok3 and Spok4, Spok3 would have to have had a much higher rate of change than both Spok1 and Spok4 in order to explain the observed divergence. In addition, the inferred deletions in the Spok3 coding sequence would have to be reverted by gene conversion with an unknown Spok homolog, and new deletions would have to appear subsequently in Spok3.
Alternatively, the diversification of the Spok genes may have been influenced by past hybridization, and we discuss a few possible scenarios here. One possibility is that Spok4 was introduced into P. comata, which then diverged to become Spok1 after the duplication event (Figure 8B). This divergence would then have to be followed by gene conversion in order to account for the shared frameshift mutation in Spok3 and Spok4. On the other hand, Spok1 may have been transferred to P. anserina (Figure 8C), and then duplicated to form Spok3 and Spok4, but this scenario requires the same additional steps as the orthology scenario. A final option is that Spok3 and Spok4 are not homologs formed via duplication but rather orthologs that evolved independently in separate populations (Figure 8D). Their current positions in the Spok block could be due to the fusion of ancestral blocks, followed by gene conversion. In P. pauciseta (CBS237.71), Spok3, Spok4, and the Spok block are nearly identical to the same sequences in P. anserina, so in order to explain the current pattern of the Spok genes and the Spok block without interspecies transfer, the block would have had to remain virtually unchanged since the divergence of P. anserina and P. pauciseta. This scenario seems highly unlikely given the relative divergence between the three species. The broader Spok homolog phylogeny presented here is also not consistent with a simple vertical decent model, supporting the conclusion that the evolution of the Spok genes has involved inter-lineage transfers. Such interspecies interactions that mediate the introgression of meiotic drive genes between species would not be a phenomenon that is unique to the Spok genes of Podospora, as meiotic drive genes in Drosophila have been observed to cross species boundaries and erode barriers of reproduction (Meiklejohn et al., 2018). Further analyses of the genomes of populations of multiple Podospora species is needed in order to resolve the history of the Spok genes and the block.
Figure 8
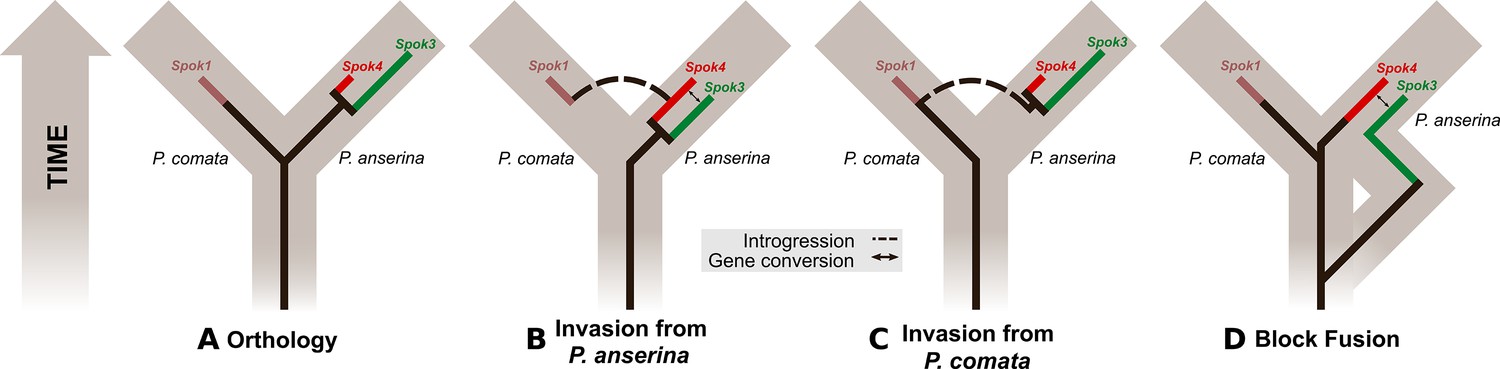
Evolutionary models of Spok diversification.
See main text for a description of the models. The models assume that an ancestral Spok homolog was present in the ancestor of both P. anserina and P. comata. The Spok diversification is traced by the inner lines. Spok2 is not included because its genomic location and sequence provide little clue as to its evolutionary history with respect to the other Spok homologs. Brown boxes represent divergence among the lineages, eventually forming P. comata and P. anserina. P. pauciseta is excluded for simplicity as in all cases it would need to obtain Spok3, Spok4 and the Spok block prior to diverging from P. anserina, and then have no divergence since the species split, or the Spok block would have to be transferred between the species.
Evolutionary history of the Spok gene family
Grognet et al. (2014) demonstrated that proteins that are related to the SPOKs are distributed across a diverse group of Ascomycota, but the majority of them are very diverged. Here, we have identified a group of more closely related homologs (clade II) from genome sequences that have been released since the Grognet et al. (2014) study, allowing us to analyze the evolutionary history at a finer resolution. The phylogenetic distribution of the clade II Spok homologs supports the general hypothesis that the Spok genes are transferred horizontally among evolutionarily disparate groups, as suggested by Grognet et al. (2014). For example, the eurotiomycete Polytolypa hystricis possesses a homolog that is closely related to the Podospora Spok genes. However, the phylogeny presented here shows that a subset of the clade II homologs agree with the relationships among closely related species (Maharachchikumbura et al., 2015), suggesting an alternative hypothesis whereby the Spok genes are ancestral to the Sordariomycetes but lost frequently. Such a scenario would imply that there are long-term consequences of possessing spore-killer genes, even if they are fixed in the population. These two hypotheses are not mutually exclusive, and with our data, we are not able to disentangle their relative importance for the observed pattern.
The diversification pattern may also give insight into the possibility that SPOK homologs function as drivers in lineages other than Podospora. The phylogeny presented here suggests that the clade I homologs do not represent meiotic drive genes because only one presumably orthologous copy is typically found. By contrast, the numerous closely related Spok homologs of clade II may be driving. For example, in F. oxysporum f. sp. cepae four nearly identical copies are found, resembling the distribution of the Spok genes in Podospora. However, no sexual cycle has been observed in F. oxysporum. Given that we demonstrate vegetative killing with Spok3, it is possible that the Fusarium Spok genes operate in vegetative tissue to ensure the maintenance of the pathogenic-associated chromosomes. Alternatively, as F. oxysporum strains have been found with both mating type alleles (O'Donnell et al., 2004), there may be a cryptic sexual cycle in which the Spok homologs are active.
Conclusions
With this study, we have provided a robust connection between the phenotype and genotype of spore-killing in P. anserina. We showed that meiotic drive in Podospora spp. is governed by genes of the Spok family, a single locus drive system that confers both killing and resistance within a single protein, which synergize to create hierarchical dynamics by the combination of homologs at different genomic locations. We define Psk-1, Psk-2, Psk-5, Psk-7, Psk-8, and Psk-S in terms of Spok gene content and describe the interactions among them. The Spok genes are prone to duplication, diversification and movement in the genome. Furthermore, our results indicate that they probably evolved via cross-species transfer, highlighting the potential risks of the release of synthetic gene drivers for biological control invading non-target species. Moreover, we present evidence that homologs of the Spok genes might have similar dynamics across other groups of fungi, including pathogenic strains of Fusarium. Taken together, the Spok system provides insight into how the genome can harbor numerous independent elements that enact their own agendas and affect the evolution of multiple taxa.
Materials and methods
Fungal material
Request a detailed protocolThe fungal strains used in this study are listed in Table 1 and were obtained from the collection maintained at the Laboratory of Genetics at Wageningen University (van der Gaag et al., 2000) and the University of Bordeaux. Strains with the ‘Wa’ identifier were collected from the area around Wageningen between 1991 and 2000 (van der Gaag et al., 1998; van der Gaag et al., 2000; Hermanns et al., 1995). Strains S, Y, and Z were collected in France in 1937 (Rizet, 1952; Belcour et al., 1997). Strain S is commonly used as a wildtype reference, and an annotated genome (Espagne et al., 2008) is publicly available at the Joint Genome Institute MycoCosm website (https://genome.jgi.doe.gov/programs/fungi/index.jsf) as ‘Podan2’. A strain labeled T (referred to herein as TG) was kindly provided by Andrea Hamann and Heinz Osiewacz from the Goethe University Frankfurt and originates from the laboratory of Denise Marcou. However, as the genome sequence of TG did not match that reported by Silar et al. (2019), but instead is a strain of P. anserina, we included in our dataset another strain labeled T from the Wageningen Collection that was originally provided by the laboratory of Léon Belcour. We referred to this strain as TD, and sequenced it using only Illumina HiSeq (see Appendix 2 for further discussion). It remains unclear where exactly TD and TG were collected, given the labeling confusion.
Representative strains of the Psk spore-killer types from the Wageningen collection were phenotyped to confirm the interactions described by van der Gaag et al. (2000). Strains Wa87 and Wa53 were selected as representative of the Psk-1 type, Wa28 for Psk-2, Wa21 for Psk-3, Wa46 for Psk-4, Y for Psk-5, Wa47 for Psk-6, and Wa58 for Psk-7. Strains S and Wa63 were used as reference strains and are annotated as Psk-S. Strain Wa58 mated poorly in general, so strain Z was also used as a mating tester for the Psk-7 spore-killer type. For all crossing experiments and genome sequencing, we isolated self-sterile monokaryons (i.e., haploid strains containing only one nuclear type) from spontaneously produced five-spored asci (Rizet and Engelmann, 1949), identified their mating type (mat+ or mat–) by crossing them to tester strains, and annotated them with + or – signs accordingly.
Culture and crossing conditions
Request a detailed protocolAll crosses were performed on Petri dishes with Henks Perfect barrage medium (HPM). This media is a modified recipe of PASM2 agar (van Diepeningen et al., 2008), to which 5 g/L of dried horse dung is added prior to autoclaving. Strains were first grown on solid minimal medium, PASM0.2. For each cross, a small area of mycelium of each of two monokaryons was excised from the plates and transferred to HPM. Perithecia (fruiting bodies) form at the interface between sexually compatible mat+ and mat– monokaryons. Mature perithecia with fully developed ascospores were harvested after 8–11 days and the percentage of two-spored asci was evaluated to determine the killing percentage (Box 1—figure 1). All cultures were incubated at 27°C under 70% humidity for a 12:12 light:dark cycle. Barrage formation, whereby confrontations between mycelia of two different strains will produce a visible line of dead cells if they are vegetatively incompatible, was also evaluated on HPM. For details, see van der Gaag et al. (2003).
Experimental design for crosses
Request a detailed protocolTo determine the epistatic interactions between the different Psks, crosses were set up according to the following design (Figure 4—figure supplement 4A). Monokaryons of two parental strains (P1 and P2) were confronted on Petri dishes with solid HPM media and perithecia were dissected upon maturation, which takes place after 9–12 days. If only four-spored asci were observed, P1 and P2 are the same Psk, otherwise they represent different Psks. A spore was selected from a two-spored ascus to generate an F1 strain for further crosses: selfing or backcrossing to the parental strains. As most F1 strains from two-spored asci will be homokaryotic for a driver, they will result in four-spored asci when selfed, except in the case of mutual killing (Figure 4—figure supplement 4B). Mutual killing can also result in completely empty asci if the drivers are at the same locus. By crossing the F1 strains to both + and – strains of P1 and P2, we can distinguish between mutual killing, mutual resistance, and dominance. If none of the crosses yield two-spored asci, there is mutual resistance (Figure 4—figure supplement 4). In a dominance interaction, for example when P1 is dominant to P2, the F1 strain will produce four-spored asci with both mating types (+/–) of P1, but will have two-spored asci with both mating types of P2. If two-spored asci were observed in crosses to both P1 and P2, or if there are two-spored asci when the F1 is selfed, then there is mutual killing.
DNA and RNA extraction and sequencing
Culturing, extracting and sequencing genomic DNA using Illumina HiSeq
Request a detailed protocolMonokaryotic strains of P. anserina were grown on plates of PASM0.2 covered with cellophane. The fungal material was harvested by scraping mycelium from the surface of the cellophane and placing 80–100 mg of mycelium in 1.5 ml Eppendorf tubes, which were then stored at −20°C. Whole-genome DNA was extracted using the Fungal/Bacterial Microprep kit (Zymo; www.zymo.com) and sequenced using the SNP and SEQ Technology platform (SciLifeLab, Uppsala, Sweden), where paired-end libraries were prepared and sequenced with the Illumina HiSeq 2500 platform (125-bp-long reads) or HiSeq X (150-bp-long reads) (Table 1).
Culturing, extracting and sequencing genomic DNA using PacBio RSII
Request a detailed protocolIn order to generate high-molecular-weight DNA that is suitable for sequencing using PacBio, eight strains were grown on PASM0.2 for 5–7 days (Table 1). The agar with mycelium was cut into small pieces and used as inoculum for flasks containing 200 ml 3% malt extract solution, which were then incubated on a shaker for 10–14 days at 27°C. The mycelia were filtered from the flasks, cut into small pieces and ~1 g was allotted into 2 ml tubes with screw-on caps, after which the tubes were stored at −20°C. High-molecular-weight DNA was then extracted following the procedure described in Sun et al. (2017). In brief, the mycelium was freeze-dried and then macerated, and DNA was extracted using Genomic Tip G-500 columns (Qiagen) and cleaned using the PowerClean DNA Clean-Up kit (MoBio Labs). The cleaned DNA was sequenced at the Uppsala Genome Center (SciLifeLab, Uppsala, Sweden) using the PacBio RSII platform (Pacific Biosciences). For each sample, 10 kb libraries were prepared and sequenced using four SMRT cells and the C4 chemistry with P6 polymerase.
Culturing, extracting and sequencing genomic DNA using MinION Oxford Nanopore
Request a detailed protocolDNA extraction was performed as for the PacBio sequencing, except that the mycelia were dissected to remove the original agar inocula and the DNA was purified using magnetic beads (SpeedBeads, GE) then sequenced without further size-selection. Monokaryotic samples TG+ and CBS237.71– were sequenced first in a barcoded run on a R9.5.1 flowcell using the Oxford Nanopore Technologies (ONT) rapid barcoding kit (1.5 µl RBK004 enzyme to 8.5 µl DNA per reaction). Owing to low tagmentation efficiency, we did additional sequencing for TG+ using the ligation sequencing kit (LSK108, R9.4.1 flowcell). 500 ng DNA (25 µl) was mixed with 1.5 µl NEB Ultra-II EP enzyme and 3.5 µl NEB Ultra-II EP buffer and incubated for 10 min at 20°C and 10 min at 65°C, before addition of 20 µl AMX adaptor, 1 µl ligation enhancer, and 40 µl NEB Ultra-II ligase. After ligation, the standard ONT washing and library loading protocol was followed and the sample was sequenced on a R9.4.1 flowcell. After sufficient sequencing depth had been achieved for sample TG+, the flowcell was washed and the remaining barcoded samples were loaded to improve coverage for sample CBS237.71–. The sample Y+ yielded less DNA (150 ng in 15 µl) and hence half the normal volume of adaptor was used (10 µl) and ligated using 20 µl Blunt/TA ligase for 15 min. Otherwise, the standard protocol was followed, with sequencing done in a R9.4.1 flowcell. Basecalling and barcode split was done using Guppy 1.6 and Porechop (ONT) for all samples.
RNA sequencing
Request a detailed protocolWe generated transcriptomic data from dikaryotic strains that undergo spore-killing during selfing. The S14 backcrosses (see below) were mated to the strain S in order to obtain killer heteroallelic spores (from four-spore asci) that were dissected from ripe fruiting bodies (see Figure 2—figure supplement 2). The spores were germinated in plates of PASM2 with 5 g/L ammonium acetate added. Two days after germination, the culture was stored in PASM0.2 media at 4°C to arrest growth. From that stock, we inoculated HPM plates with either a polycarbonate track etched 76 mm 0.1 µm membrane disk (Poretics, GVS Life Sciences, USA) (Psk1xS5 and Psk7xS5) or a cellophane layer (Psk2xS5 and Psk5xS5) on top. The mycelia were grown for ~11 days and harvested for RNA extraction when the first spores were shot into the plate lid, ensuring several stages of fruiting body development. Note that P. anserina starts to degrade cellophane after ~6 days, and therefore the polycarbonate membrane allows for longer growing periods. Spore-killing was independently confirmed on HPM plates inoculated without a membrane. In addition, in order to improve gene annotation, we grew the strains Wa63– and TD+ on a cellophane layer on HPM for 11 and 7 days, respectively, to capture transcripts occurring during the monokaryotic phase.
The harvested mycelia were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction. Next, 150 mg of frozen tissue was ground under liquid nitrogen and total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany). The quality of RNA was checked on the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). All RNA samples were treated with DNaseI (Thermo Scientific). Sequencing libraries were prepared using an NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs). The mRNA was selected by purifying polyA+ transcripts (NEBNext Poly(A) mRNA Magnetic Isolation Module, New England Biolabs). Finally, paired-end libraries were sequenced with Illumina HiSeq 2500 at the SNP and SEQ Technology platform.
Genomic analyses
Request a detailed protocolFor both DNA and RNA Illumina HiSeq reads, adapters were identified with cutadapt v. 1.13 (Martin, 2011) and then trimmed using Trimmomatic 0.36 (Bolger et al., 2014) with the options ILLUMINACLIP:adapters.fasta:1:30:9 LEADING:20 TRAILING:20 SLIDINGWINDOW:4:20 MINLEN:30. Only filtered reads with both forward and reverse were kept for downstream analyses. For short-read mapping, we used BWA v. 0.7.17 (Li and Durbin, 2010) with PCR duplicate marking of Picard v. 2.18.11 (http://broadinstitute.github.io/picard/), followed by local indel re-aligning implemented in the Genome Analysis Toolkit (GATK) v. 3.7 (Van der Auwera et al., 2013). Mean depth of coverage was calculated with QualiMap v.2.2 (Okonechnikov et al., 2016).
The raw PacBio reads were filtered and assembled with the SMRT Analysis package and the HGAP 3.0 assembler (Chin et al., 2013). The resulting assembly was error-corrected (polished) with Pilon v. 1.17 (Walker et al., 2014) using the mapped filtered Illumina reads of the same monokaryotic strain. The samples sequenced with MinION were assembled using Minimap2 v. 2.11 (Li, 2018) and Miniasm v. 0.2 (Li, 2018; Li, 2016), polished twice with Racon v. 1.3.1 (Vaser et al., 2017) using the MinION reads, and further polished for five consecutive rounds of Pilon v. 1.22 using the Illumina reads as above. Scaffolds were assigned to chromosome numbers on the basis of homology with Podan2. Small scaffolds (<100 kb) corresponding to rDNA and mitochondrial-derivatives were discarded. Only the biggest mitochondrial scaffold was retained. In addition, DNA Illumina reads were assembled de novo for each sample using SPAdes v. 3.12.0 (Bankevich et al., 2012; Antipov et al., 2016) using the k-mers 21,33,55,77 and the –careful option. BLAST searches of the scaffolds in the final assembly of the strain CBS237.71 revealed contamination by a Methylobacterium sp. in the MinION data (but not in the Illumina data set). The scaffolds matching the bacterium were removed from the analysis. Long-read assemblies were evaluated using BUSCO v. 3.0.2 (Simão et al., 2015; Waterhouse et al., 2017) for the Sordariomyceta ortholog set with the following dependencies: BLAST suit 2.6.0+ (Camacho et al., 2009), HMMER v, 3.1b2 (Mistry et al., 2013), and AUGUSTUS v 3.2.3 (Stanke and Waack, 2003). Short-read assemblies were evaluated using QUAST v. 4.6.3 (Mikheenko et al., 2016).
The assembly of the Spok block was visually inspected by mapping the long reads (using Minimap2) and the short reads (BWA) as above into the long-read polished assemblies. As the MinION assemblies maintain some degree of sequencing error at repetitive regions that cannot be confidentially polished, we also assembled both types of reads into a hybrid assembly using SPAdes (same options as above) and, whenever different for short indels or SNPs but fully assembled, the sequence of the Spok genes was taken from the (low-error) hybrid assembly. Assembly of the Spok block of the TG+ strain was particularly challenging because the recovered MinION reads were relatively short. However, a few (<10) reads were long enough to cover the tandem duplication that contains Spok3 (albeit with high nucleotide error rate in the assembly). The hybrid SPAdes assembly collapsed the duplication into a single copy. We therefore mapped the short reads into the hybrid assembly, confirming that the Spok3 gene had doubled coverage and no SNPs, as expected for a perfect duplication.
Alignments of the assembled genomes were performed with the NUCmer program of the MUMmer package v. 4.0.0beta2 (Kurtz et al., 2004) using options –b 200 c 2000 –maxmatch, except when otherwise noted. The figures showing alignments of chromosome 5, the Spok block, and the Spok2 region (Figure 3, Figure 5, and Figure 5—figure supplements 1 and 2) were generated by extracting the regions from each de novo assembly and aligning them in a pairwise fashion. The NUCmer output was then visualized using a custom Python script. The distribution of GC was plotted along the chromosomes with a custom Python script using 4-kb windows and steps of 2 kb.
In order to evaluate synteny across species, we aligned the chromosomal scaffolds of Wa58– (P. anserina, Psk-7), CBS237.71- (P. pauciseta), and the reference genome of P. comata (PODCO, Silar et al., 2019) using NUCmer with –b 2000 in an all-vs-all fashion. Given that the largest TE reported for P. anserina is around 12 kb (Espagne et al., 2008), we filtered out alignments smaller than 13 kb and plotted the remaining alignments using Circos (Krzywinski et al., 2009). We further excluded alignments of missing data (Ns) tracks in chromosome 6 and 7 from PODCO. See https://github.com/johannessonlab/SpokPaper (Ament-Velásquez, 2019; copy archived at https://github.com/elifesciences-publications/SpokPaper) for a Snakemake pipeline and Circos configuration files. Notice that the Circos plot includes intrachromosomal alignments; for example, the rDNA operon in chromosome 3 is especially noticeable for Wa58–. To evaluate the large-scale translocations in P. comata (Figure 1B and Figure 1—figure supplement 1), we mapped the short-reads of TD+ to PODCO and Podan2, inferring mis-assemblies on the basis of the concordance of paired-end reads. Translocation 1 is clearly a misassembly. The translocation 2 in chromosome 4 however is complex because the corresponding boundaries in Podan2 start at a cluster of TEs at the 5′ end, and finish at the centromere on the 3′ end. Indeed, Silar et al. (2019) could not verify this translocation using PCR. Accordingly, the mapping of the paired-end reads does not support the translocation to the end of chromosome 4 in PODCO.
Genome annotation
Request a detailed protocolFor annotation, we opted for gene prediction trained specifically on P. anserina genome features. We used the ab initio gene prediction programs GeneMark-ES v. 4.32 (Lomsadze et al., 2005; Ter-Hovhannisyan et al., 2008) and SNAP release 2013-06-16 (Korf, 2004). All of the training process was performed on the sample Wa28–, for which all chromosomes were assembled (see 'Results'). The program GeneMark-ES was self-trained with the script gmes_petap.pl and the options –fungal –max_intron 3000 min_gene_prediction 120. SNAP was trained as instructed in the tutorial of the MAKER pipeline v. 2.31.8 (Holt and Yandell, 2011; Campbell et al., 2014, and in the SNAP README file). First, we use the Podan2 transcripts and protein models as sole evidence to infer genes with MAKER (option est2genome = 1) and then we had a first round of SNAP training. The resulting HMM file was used to re-run MAKER (est2genome = 0) and to re-train SNAP, obtaining the final HMM training files.
A library of repetitive elements was constructed by collecting the reference P. anserina TEs described in Espagne et al. (2008) available in Genbank, and combining them with the fungal portion of Repbase version 20170127 (Bao et al., 2015) and the Neurospora library of Gioti et al. (2013). In order to produce transcript models, we used STAR v. 2.6.1b (Dobin et al., 2013) with maximum intron length of 1000 bp to map the RNAseq reads of all samples, followed by processing with Cufflinks v. 2.2.1 (Trapnell et al., 2010). For the final genome annotation, we used MAKER v. 3.01.02 along with GeneMark-ES v. 4.33, SNAP release 2013-11-29, RepeatMasker v. 4.0.7 (http://www.repeatmasker.org/), BLAST suit 2.6.0+ (Camacho et al., 2009), Exonerate v. 2.2.0 (Slater and Birney, 2005), and tRNAscan-SE v. 1.3.1 (Lowe and Eddy, 1997). After preliminary testing, we chose the transcripts of Psk7xS14 (mapped to the PacBio assembly of Wa58–) and Wa63– (PacBio assembly of the same strain) as expressed sequence tag (EST) evidence, and the Podan2 and TD (Silar et al., 2019) models as protein evidence. The MAKER models of relevant regions were manually curated by comparing with RNAseq mapping and coding sequences (CDS) produced with TransDecoder v. 5.5.0 (Haas et al., 2013) on the Cufflinks models.
We used the blastn program to localize possible copies of Spok genes in all genome assemblies. The Spok2 (Pa_5_10) gene from Grognet et al. (2014) was selected as the query. We named the new Spok genes (Spok3 and Spok4) arbitrarily on the basis of sequence similarity, as reflected in the phylogenetic analyses (see below). Note that the existence of Spok3 had previously been hypothesized by Grognet et al. (2014), but no DNA sequence was provided. Moreover, the strain Y, in which they identified Spok3, contains both Spok3 and Spok4.
Introgressions of the spore-killing loci
Request a detailed protocolBackcrossed strains of the various spore-killer phenotypes were generated through five recurrent backcrosses to the reference strain S (S5) by van der Gaag et al. (2000) . In the original study, the strains selected as spore-killer parents were Wa53+ for Psk-1, Wa28– for Psk-2, Y+ for Psk-5, and Wa58– for Psk-7. The S5 strains are annotated as Wa170 (Psk-1), Wa130 (Psk-2), Wa200 (Psk-5), and Wa180 (Psk-7) in the Wageningen collection, but for the sake of clarity, we refer to them as Psk1xS5, Psk2xS5, Psk5xS5, and Psk7xS5.
We sequenced the S5 strains along with the reported parental strains using Illumina HiSeq 2500. We mapped the reads to Podan2 as described above, and then performed SNP calling using the HaplotypeCaller pipeline of GATK (options: –ploidy 1 –newQual –stand_call_conf 20.0). We removed sites that had missing data, that overlapped with repeated elements as defined by RepeatMasker, or in which all samples were different from the reference genome, using VCFtools v. 0.1.16 (Danecek et al., 2011), BEDtools v. 2.27.1 (Quinlan and Hall, 2010), and BCFtools v. 1.9 (Danecek and McCarthy, 2017), respectively. We plotted the density of filtered SNPs across the genome with the R packages vcfR (Knaus and Grünwald, 2017; Kamvar et al., 2015) and poppr (Knaus and Grünwald, 2017; Kamvar et al., 2015). A full Snakemake (Johannes and Rahmann, 2018) pipeline can be found at https://github.com/johannessonlab/SpokPaper. Notice that we sequenced both monokaryons of our strain S to account for the mutations that might have had occurred since the separation of the reference S strain in the laboratory of Espagne et al. (2008) and our S strain from the Wageningen collection. These mutations should be present in the backcrosses, but they are independent from the spore=killer elements.
Inspection of the introgressed tracks revealed that the variants of the backcross Psk1xS5 do not match Wa53+ (the reported parent) perfectly. Given that the Spok content is the same as that in Wa53+, the introgressed track co-occurs with the expected position of the Spok block on chromosome 3, and the fact that the phenotype of this backcross matches a Psk-1 spore-killer type, we concluded that Wa170 (Psk1xS5) in the collection actually belongs to another of the Psk-1 backcrosses described in the doctoral thesis of van der Gaag (2005), probably backcrossed from Wa52. Puzzlingly, an introgressed track in the chromosome 3 of the Psk2xS5 strain does not match the expected parent (Wa28) either, both in SNPs and het-gene alleles Figure 4—figure supplement 3). However, other tracks in different chromosomes, including that of chromosome 5 where the Psk-2 Spok block can be found, do match Wa28. Likewise, like Wa28, Psk2xS5 only has Spok2 and Spok3 copies. Hence, we concluded that our results are not affected by these inconsistencies.
As reported by van der Gaag et al. (2000), the S5 strains were generated by selecting ascospores from two-spored asci of crosses between S and the spore killer parent. This procedure ensures that the offspring will be homozygous for alleles of the spore-killer parent from the spore-killing locus to the centromere (Box 1—figure 1 and Figure 4—figure supplement 3). To eliminate as much background as possible from the spore killer parents in the backcrossed strains, nine additional backcrosses were conducted where ascospores were selected from four-spored and two-spored asci in alternating generations. Ascospores from the final generation were selected from two-spored asci to ensure that the strains would be homozygous at the spore-killing locus. These strains are the result of 14 backcrosses to S (S14) and are annotated as Psk1xS14, Psk2xS14, Psk5xS14, and Psk7xS14. The S5 and S14 strains were phenotyped by crossing the strains to their parents as well as other reference spore-killer strains to confirm that the killing phenotypes remained unchanged after the backcrosses.
Knock-out of Spok2
Request a detailed protocolTo knock-out Spok2, a 459-bp and a 495-bp fragment flanking the Spok2 ORF downstream and upstream were obtained by PCR. These fragments were then cloned flanking the hph gene in the SKhph plasmid as blunt end fragments in a EcoRV site and a SmaI site. The deletion cassette was then amplified by PCR and used to transform a ΔKu70 strain (El-Khoury et al., 2008). Five transformants were screened for integration of the hph marker at Spok2 by PCR and crossed to s. To purify the ΔSpok2 nuclei, a heterokaryotic binucleated ΔSpok2 spore was recovered from a two-spored ascus and used to fertilize the initial ΔSpok2 transformant (which may or may not be heterokaryotic). Uninucleated hygR-resistant spores were then recovered from this cross.
Construction of a disruption cassette to insert Spok3 or Spok4 into the PaPKS1 locus
Request a detailed protocolTo replace the ORF of the centromere-linked Pa_2_510 (PaPKS1) gene by one of the Spok3 or Spok4 genes (see 'Results'), a disruption cassette was constructed as follows. A DNA fragment corresponding to the 700-bp upstream region of the PaPKS1 ORF was amplified with oligonucleotides UpPks1_F and UpPks1_R. This fragment was then cloned into a SKpBluescript vector (Stratagene) containing the nourseothricin-resistance gene Nat in the EcoRV site (vector named P1) using the SacII/NotI restriction enzymes (upstream from the Nat gene) to produce the P1UpstreamPKS1 vector. Then, the 770 bp downstream of the PaPKS1 ORF was amplified with oligonucleotides DownPks1_F and DownPks1_R. This second fragment was cloned into the P1UpstreamPKS1 vector using the HindIII/SalI restriction enzymes (downstream from the Nat gene) to produce the P1UpstreamDownstreamPKS1 vector. Finally, Spok3 was amplified from the Wa28 strain with oligonucleotides UpSpok3 and 4_F and DownSpok3, and Spok4 was amplified from the Wa87 strain with oligonucleotides UpSpok3 and 4_F and DownSpok4. These genes were then cloned into the P1UpstreamDownstreamPKS1 vector using the NotI/XbaI restriction enzymes (between PaPKS1 upstream region and Nat gene) to produce the P1UpstreamDownstreamPKS1_Spok3 or the P1UpstreamDownstreamPKS1_Spok4 vector, so that the Spok3or Spok4 and Nat genes are flanked by the upstream and downstream regions of PaPKS1 ORF, allowing PaPKS1 ORF replacement by homologous recombination. The Spok3 and Spok4 amplified Spok genes contain the ORFs flanked by the 983-bp upstream the start codon and the 460-bp downstream the stop codon for Spok3, and by the 984-bp upstream the start codon and the 393-bp downstream the stop codon for Spok4, allowing the expression of the Spok genes using their native promoter and terminator regions. The disruption cassettes were then amplified from the final vectors using the most distal oligonucleotides 3′insidePsk1_F and 3′insidePsk1_R and named PKS1::Spok3_nat-1 and PKS1::Spok4_nat-1. See Supplementary file 4 for primer sequences. Point mutations in the Spok3 gene were obtained by site-directed mutagenesis using the Q5 high-fidelity polymerase (New England Biolabs) and verified by Sanger sequencing.
The P. anserina ΔSpok2 (ΔPa_5_10) strain was obtained after disruption of the gene Pa_5_10 and replacement of its ORF with the hygromycin-resistance gene hph in a ΔKu70 strain. This strain was used as recipient strain for the disruption cassettes. We used 5 µl of the cassettes for transfection and Nourseothricin-resistant transformants were selected. As expected, most of the transformants were unpigmented and corresponded to the insertion of Spok3 or Spok4 by replacement of PaPKS1. Gene replacement was verified by PCR.
Protein-annotation methods
Request a detailed protocolPrediction of unstructured regions was performed in SPOK3 with PrDOS with a 2% false-positive setting (Ishida and Kinoshita, 2007). Coiled-coil prediction was performed with LOGICOIL (Vincent et al., 2013), CCHMM_PROF (Bartoli et al., 2009) and Multicoil2 (Wolf et al., 1997). Domain prediction was performed using Gremlin (Balakrishnan et al., 2011) and RaptorX contact predict (Ma et al., 2015). Conserved residues were identified using Weblogo 3 (Crooks et al., 2004) with a Gremlin-generated alignment as input. Domain identification was done with HHPred (Zimmermann et al., 2018).
In order to compare the diversity at the nucleotide level with the protein models, we calculated the average pairwise nucleotide differences (Nei and Li, 1979) for each bi-allelic site (correcting by the number of sites (n/(n –1)) while ignoring sites with gaps) on a Spok alignment (see below), using overlapping windows of 100 bp and steps of 20 bp. This procedure was performed on a selected representative of each Spok homolog (Spok2 of S, Spok3 and Spok4 of Wa87, and Spok1 from TD), or for all the alleles of each Spok within the P. anserina strains. Values of dN/ds were calculated using the seqinr package in R (Charif and Lobry, 2007).
In order to infer the relationship between the three Podospora taxa investigated herein, we used OrthoFinder v. 2.2.6 (Emms and Kelly, 2015; Emms and Kelly, 2019) to define orthogroups across all samples with long-read data and the reference genomes of P. anserina (Espagne et al., 2008) and P. comata (Silar et al., 2019). We randomly selected 1000 orthogroups of single-copy orthologs. As OrthoFinder works with protein sequence, we used the Podan2 ortholog to do BLAST searches against each genome and extracted the corresponding best hit as nucleotide sequence (including introns). We then aligned each orthogroup using MAFFT v. 7.407 (Katoh et al., 2017) with options ––maxiterate 1000 –retree 1 –localpair. We concatenated the 1000 alignments (1,652,987 sites, from which 27,000 were variable) and used the resulting matrix as input for SplitsTree4 v. 4.14.16, build 26 Sep 2017 (Huson and Bryant, 2006) to construct an unrooted split network with a NeighborNet (Bryant and Moulton, 2002) distance transformation (uncorrected distances) and an EqualAngle splits transformation. To estimate the genetic (gene) distance between species, we averaged the identity across all P. anserina samples vs P. pauciseta (CBS237.71) and vs. P. comata (Silar et al., 2019), using the values produced in the distance matrix of SplitsTree.
The final gene models of all the Spok genes in Podospora spp. were aligned along with the sequences of Spok2 and Spok1 from Grognet et al. (2014) using MAFFT online version 7 (Katoh et al., 2017) with default settings (only one copy of Spok3 from TG was used). The resulting alignment was manually corrected taking into account the reading frame of the protein. As the UTRs seem to be conserved between paralogs, 654 (5' end) and 250 (3' end) bp of the flanking regions with respect to Spok2 were also included in the alignment. An unrooted split network was constructed in SplitsTree4 as above. SplitsTree4 was used likewise to perform a Phi test for recombination (Bruen et al., 2006), using a windows size of 100 and k = 6. In addition, we used the BlackBox of RAxML-NG v. 0.6.0 (Kozlov et al., 2019) to infer maximum likelihood phylogenetic trees of the nucleotide alignment of the 5' UTR, the coding sequence (CDS), and the 3' UTR of the Spok homologs. We ran RAxML-NG with 10 parsimony and 10 random starting trees, a GTR +GAMMA (four categories) substitution model, and 100 bootstrap pseudo-replicates for each analysis.
In order to create a phylogeny of proteins that are closely related to the products of the Spok genes in Podospora (and hence likely to be meiotic drivers), the protein sequence of Spok1 was used as a query against the NCBI genome database (as of 14 November 2018). We collated all hits with e-values lower than Fs_82228, which has been shown previously to have some spore-killing functionality in P. anserina (Grognet et al., 2014), with hit coverage greater than 75% and no missing data (Ns) in the sequence. The sequences were aligned using the codon-aware program MACSE v. 2.03 (Ranwez et al., 2018), with the representative Podospora Spok genes set as ‘reliable’ sequences (–seq), and the rest as ‘non reliable’ (–seq_lr). Many of the original gene models predict introns in the sequences, but no divergent regions were apparent in the alignment and, even if present, MACSE tends to introduce compensatory frame shifts. The entire gene alignment was used for the analysis. The resulting nucleotide alignment was corrected manually, translated into amino acids, and trimmed with TrimAl v. 1.4.1 (Capella-Gutiérrez et al., 2009) using the gappyout function. A maximum likelihood tree was then produced using IQ-TREE v. 1.6.8 (Kalyaanamoorthy et al., 2017; Nguyen et al., 2014) with extended model selection (–m MFP) and 1000 standard bootstrap pseudo-replicates. The protein sequence Uv_5543 of Ustilaginoidea virens was selected as an outgroup on the basis of a BioNJ tree made with SeaView v. 4.5.4 (Gouy et al., 2010) of the Gremlin alignment described above.
For comparison, we performed a phylogenomic analysis of all of the strains that had at least one BLAST hit for the Podospora Spok homologs, as defined above. Briefly, we recovered all the protein sequences for each genome from GenBank and ran OrthoFinder to recover orthogroups. We obtained 288 single-copy orthogroups that were then processed with PREQUAL v.1.02 (Whelan et al., 2018) and aligned with MAFFT. Columns with more than 50% missing data were removed with TrimAl (–gt 0.5) and all alignments were concatenated. The supermatrix was analyzed with IQTree as above but with 100 standard bootstraps.
Pool-sequencing of Psk-1 vs Psk-5 progeny
Request a detailed protocolIn order to confirm that Spok2 is responsible of the killing relationship between Psk-5 and Psk-1, we conducted a cross between the strains Wa87 and Y. When perithecia started shooting spores, we replaced the lid of the cross plate with a water-agar plate upside-down, and let it sit for around an hour. As the P. anserina spores from a single ascus typically land together, it is possible to distinguish spores that came from an ascus with no killing (groups of four spores) from those that survived killing (groups of two spores). To improve germination rates, we scooped spore groups of the same ascus type and deposited them together in a single plate of germination medium. After colonies became visible, they were transferred into a PASM2 plate with a cellophane layer where they grew until DNA extraction, which was followed by pool-sequencing with Illumina HiSeq X. In total, 21 two-spore groups, and 63 four-spore groups were recovered.
The resulting short reads were quality controlled and mapped to Podan2 as above. We used GATK to call variants from the parental strains (treated as haploid) and the two pool-sequencing databases (as diploids). We then extracted SNPs, removed sites with missing data, and attempted to quantify the coverage frequency of the parental genotypes for each variant. The expectation was that spore killing (two-spore asci) would result in a long track of homozygosity (only one parental genotype) around Spok2, as compared to the fully heterozygous four-spore asci. A full Snakemake pipeline is available at https://github.com/johannessonlab/SpokPaper.
Appendix 1
The biology of Podospora
The life cycle of P. anserina is an important factor to consider when discussing the meiotic drive of the Spok genes. Although it has haploid nuclei, P. anserina maintains a dikaryotic (n + n) state throughout its entire lifecycle. Haploid nuclei of different mating-types are shown as white and black circles within fungal cells. The fruiting body (perithecium) is generated from dikaryotic (n + n) mycelia, usually from a single individual strain. Within the perithecium, the sexual cycle is completed to produce four dikaryotic ascospores per ascus. Occasionally, atypical spore formation may occur and can result in the production of five spores in an ascus, of which two are small and monokaryotic (n). These are self-sterile and need to outcross either with a monokaryotic individual of the opposite mating type or with a dikaryotic individual to complete the life cycle. Note that outcrossing may occur via mating between either siblings or unrelated individuals of the opposite mating type. The monokaryotic spores are useful for generating self-sterile (haploid) cultures for the purposes of sequencing and laboratory mating and are used to generate the + and -strains that are discussed in the main text.
Appendix 1—figure 1
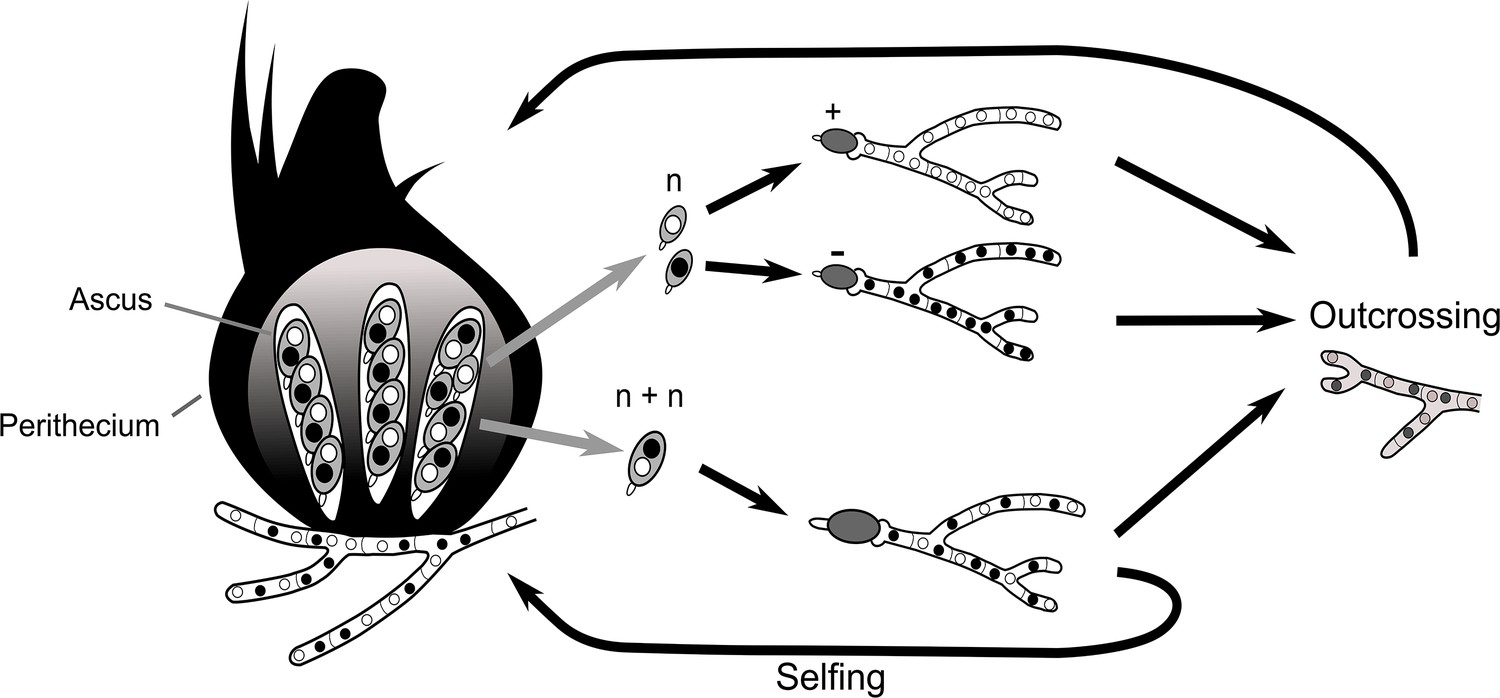
Simplified life cycle of P. anserina with explanation for isolating monokaryotic spores.
https://doi.org/10.7554/eLife.46454.040Two-locus spore-killing interaction
The interaction between Psk-1 and Psk-7 provides a good example of how the meiotic drive dynamics of P. anserina result in killing, even though both Psk-1 and Psk-7 possess the same Spok homologs (mutual resistance). The three Spok homologs (Spok2, Spok3, and Spok4) are all present in both Psk-1 and Psk-7. The observed mutual resistance is thus due to the fact that the Spok block (with Spok3 and Spok4) is located on different chromosomes. Because chromosomes segregate independently at meiosis, the expected killing percentage can be calculated as:
where 0.5 is due to independent assortment of chromosomes, fk1 is the killing percentage of strain 1, fk2 is the killing percentage of strain 2, and fsk is the spore-killing frequency observed between the two strains. For Psk-1 crossed to Psk-7, this equals 0.27. This agrees well with the observed killing percentage of 23–27% (Figure 4—figure supplement 1).
Appendix 2
History of spore-killer research in Podospora
Throughout the history of spore-killing research in Podospora, a number of observations have been made along with corresponding hypotheses. The discovery of Spok3 and Spok4 provides us with the opportunity to reinterpret these data in light of the results presented herein. Here, we will address data from four important studies: Padieu and Bernet (1967), van der Gaag et al. (2000), van der Gaag et al. (2003), and Hamann and Osiewacz (2004).
Inconsistencies among the Psk designations
Our phenotyping is in accordance with the results of van der Gaag et al. (2000) for strains Wa28, Wa53, Wa58, Wa63, Wa87, S, and Z, but contradictions were observed for Wa21, Wa46, Wa47, and Y. Strain Wa21 was previously categorized as Psk-3, which is typified by inconsistent spore killing with Psk-S strains. Here we observed stable percentages and thus consider Wa21 to be representative of Psk-2. The role of Psk-3 as a spore killer has been in doubt since its description (van der Gaag et al., 2000). This is in part due to the fact that the ascospores are not fully aborted as they are for the other spore-killer types. Instead, small transparent ascospores can still be observed within the ascus. Here, we were unable to find support for this spore-killer type and there is no clear correlation between its phenotype and any Spok genes. We therefore find it likely that the effect is due to other incompatibility factors rather than meiotic drive.
We did not observe any spore-killing in crosses between Wa46 (Psk-4) and Wa47 (Psk-6) as reported in van der Gaag et al. (2000). Two other strains had been annotated as Psk-6, Wa89 and Wa90, but no other strains were recorded as Psk-4. Unfortunately we were not able to phenotype these strains and so we are unable to evaluate Psk-6 further in this study. In addition, results from crosses of Psk-4 with a Psk-S strain (Wa63) reveals that there is a dominance interaction between them, with Psk-S killing Psk-4. This is the opposite of what was proposed in van der Gaag et al. (2000), i.e. that Psk-S kills Psk-4. Potentially, the original interpretation was hindered by poor mating of the Psk-4 strain with tester Psk-S strains. Previously, strain Y was reported to have mutual resistance with Psk-1, to be susceptible to Psk-7, and to be dominant over all other types. Here we report that Y is susceptible to Psk-1 and Psk-7, and has mutual killing with all other types, except in crosses with naïve strains where it is dominant.
Allorecognition (het) genes and spore killing
As the het-s gene is capable of causing both vegetative incompatibility and spore killing, it was hypothesized that the Psk loci may be as well. The S5 strains all demonstrate barrage formation (symptomatic of vegetative incompatibility) with strain S (van der Gaag et al., 2003). However when additional backcrosses were performed to generate S14 strains, no barrages were observed (Appendix 2—figure 1). This indicates that the spore-killing types do not directly affect vegetative incompatibility or vice versa, but may be linked to loci which do. Note that the S5 strains contain multiple genomic regions that are not isogenic with S, some of which contain known allorecognition genes (Figure 4—figure supplement 3).
Appendix 2—figure 1

Barrage tests of the S14 strains.
Strains Wa126, Wa76, Wa52, and Wa125 are wild isolates of P. anserina in the Wageningen collection. The thick white lines of mycelia demonstrate a barrage, which is indicative of heterokaryotic incompatibility in fungi. No barrages are seen among the S14 strains.
Incomplete penetrance of Spok2
To investigate the nature of the three-spored asci, tetrad dissections were conducted with asci from crosses between the Psk-S strains Wa63 and Us5, and the naïve strain Wa46. If the three-spored asci were the result of a four-spored ascus in which one of the spores aborted, all three spores should be heteroallelic for Spok2. If the three-spored asci are the result of incomplete penetrance of the killing factor, two spores should be homoallelic for Spok2 while the other spore should have no copy of Spok2. Unfortunately, spores from the crosses had very low germination rates (1/15 for Wa63 x Wa46 and 1/12 for Us5 x Wa46) as compared to other crosses (generally close to 100% germination). The progeny from the successfully germinated spores were backcrossed to the parental strains and also allowed to self to infer their Spok2 genotype. Crosses with the Wa63/Wa46 progeny revealed this strain to be homoallelic for Spok2, and crosses with the Us5/Wa46 progeny revealed it to have no copy of Spok2. Both of these observations are consistent with the hypothesis of incomplete penetrance of Spok2.
Strain T and the original reports of spore killing in Podospora
The strain T has featured prominently in a number of important publications on spore killing in Podospora. It was one of the two strains investigated in the original description of spore killing by Padieu and Bernet (1967) translated and reinterpreted by Turner and Perkins (1991), it was the strain in which Spok1 was described (Grognet et al., 2014), and it was part of an investigation of spore killing in German strains of Podospora (Hamann and Osiewacz, 2004). Our results clearly demonstrate that two strains labeled as T (TG and TD herein) are not only different strains, but are different species. The description of spore-killing in Padieu and Bernet (1967) matches our observations of crosses between TG and the Psk-S strain Wa63, including incomplete penetrance as implied by the presence of three-spored asci. Thus, we believe TG to be representative of the original T strain. In light of this, we reinterpret the results of both Padieu and Bernet (1967) and Hamann and Osiewacz (2004) as informed by the interactions of the Spok genes.
In Padieu and Bernet (1967), a cross between two strains, T and T', is described. They identify two genes (one present in T and the other in T') that cause spore-killing and interact as mutual killers. The gene from T has a killing percentage of 90%, whereas the one from T' has a killing percentage of 40% and occasionally produces three-spored asci. This fits well with a cross of Psk-5 and Psk-S, in which Psk-5 kills at 90% and Spok2 of the Psk-S strain kills at 40%, but has incomplete penetrance resulting in three-spored asci. Unfortunately, strain T' has to our knowledge not been maintained in any collections, so this cannot be confirmed experimentally. However, Psk-S strains are the most abundant phenotype from French, German, and Dutch populations (T' was isolated in France along with T) (van der Gaag et al., 2000; Grognet et al., 2014; Hamann and Osiewacz, 2004).
Hamann and Osiewacz (2004) presented a number of interesting observations. They reported a new spore-killer type, identified progeny that appeared to demonstrate gene conversion of the killer locus, and observed apparent recombinant spore-killer types. The study mostly centres around strain O, which they report to be of the same spore-killer type as TG and should thus be Psk-5 given our results. As such, we suspect that their focal cross between O and Us5, a Psk-S strain, is the same as that in the Padieu and Bernet (1967) paper. We have independently confirmed that Us5 (kindly provided by A Hamann and H Osiewacz) is Psk-S, but strain O has not been maintained in any collection. Hamann and Osiewacz (2004) also state that strain He represents a new type of spore killer. However, with O classified as Psk-5, the interactions of He match that of a Psk-1 strain. Furthermore, strain He exhibited no spore-killing with a Psk-1 strain from Wageningen. From the cross of O and Us5, they identify a number of progeny with unexpected genotypes. They interpret these genotypes as evidence of both gene conversion and recombinant spore killer types. However, under a two-locus model of mutual killing, both effects can be explained by incomplete penetrance of Spok2 (Appendix 2—figure 2). As the cross with Us5 showed a particularly high degree of anomalous results, it is possible that Us5 contains a unique allele of Spok2 that is a particularly weak killer.
Appendix 2—figure 2
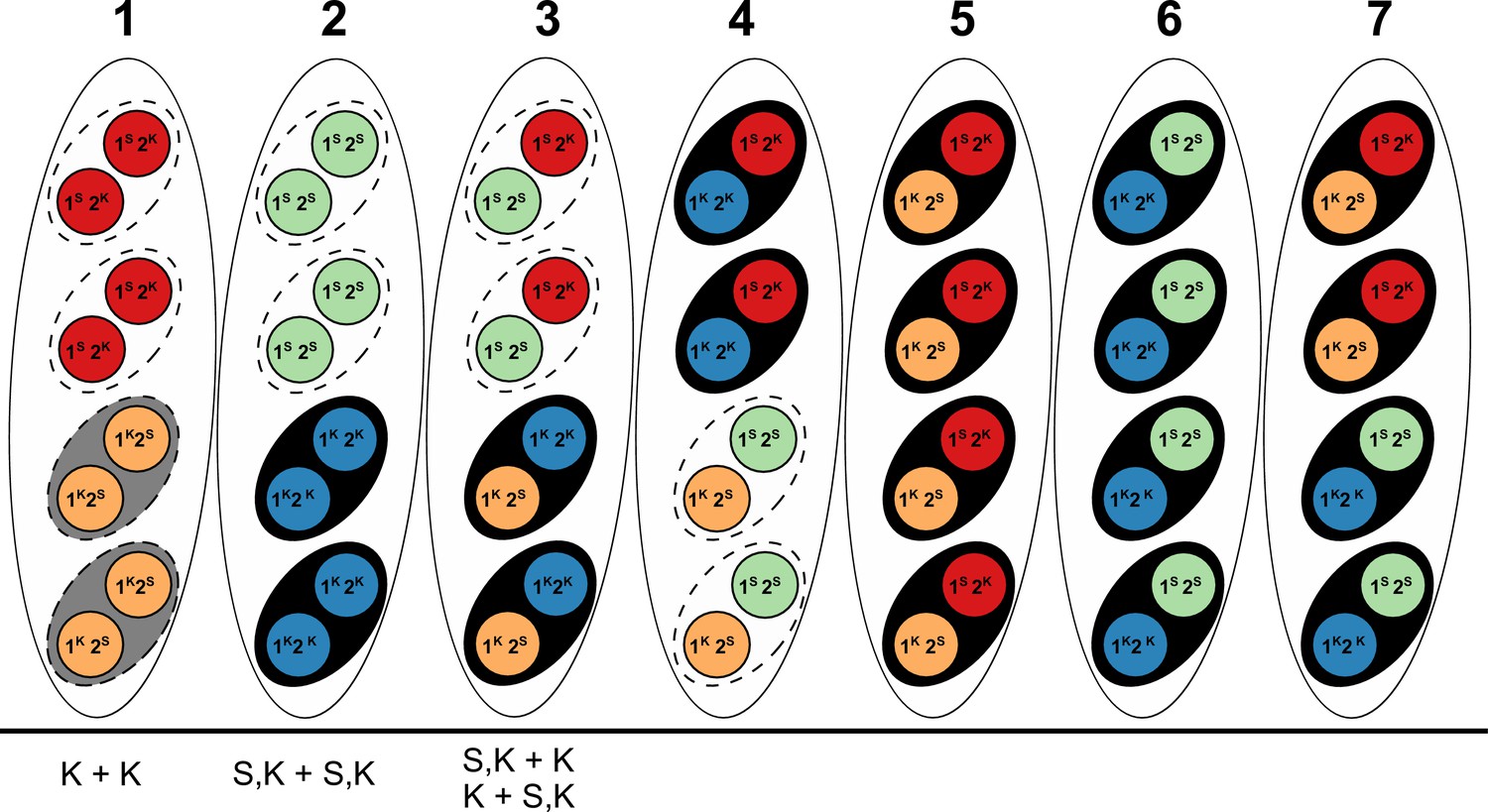
Explanation of results from a (presumably) Psk-5 vs Psk-S cross in Hamann and Osiewacz (2004) with information about Spok genes as described in the text.
The seven asci represent the possible genotype combinations of a cross between a Psk-5 strain and a Psk-S, as illustrated in Turner and Perkins (1991). Black ovals represent the ascospores, dashed ovals represent killed spores, and colored circles represent the individual nuclei, with each color corresponding to a given genotype. Genotypes are annotated as in Turner and Perkins (1991), wherein locus 1 corresponds to a killer locus with 90% FDS, the Psk-5 Spok block, and locus 2 represents a killer locus with 40% FDS, Spok2. Red nuclei represent the Psk-S parental genotype with Spok2, orange nuclei represent the Psk-5 parental genotype with Spok3 and Spok4, green nuclei represent the recombinant genotype with no Spok genes, and blue nuclei represent the recombinant genotype with Spok2, Spok3, and Spok4. Note that Spok3 and Spok4 are linked and do not segregate independently. Given its Spok content, the ascus type 1 should experience mutual killing, resulting in the abortion of all spores. Ascus types 2 to 4 should contain only two surviving spores, whereas ascus types 5 to 7 should have all spores surviving. Below, the asci are our interpretations of the annotations from Hamann and Osiewacz (2004). In their study, these authors isolated F1 spores from four-spore asci, genotyped them on the basis of crosses to the parental strains, and found no spore-killing after selfing in a few cases. In their terminology, K + K strains would correspond to a strain with the Psk-5 (grey spore) parental genotype of ascus type 1. This type of ascus should produce empty asci, so the fact that they are observed from four-spored asci suggests that when mutual killing occurs, the four spores may still develop. However, as no S + S strains (i.e. spores with two red nuclei in the type ascus 1) were reported, we can infer that only the Psk-5 type (grey spore) may be viable. S,K + S,K strains are not indicative of a recombinant killer locus as suggested in the original work, but represent strains with all three Spok genes as produced in ascus type 2. The FDS frequencies reported suggest that the isolated strains are indicative of the blue nuclear genotype and not the green nuclear genotype. Hence, the spores containing only green nuclei might not be viable either. The S,K + K and K + S,K strains are indistinguishable from each other and are indicative of the surviving spores of a type 3 ascus. These strains should exhibit spore killing when selfed because of the distribution of Spok2. However, cases in which spore killing was not observed in their study could be explained by incomplete penetrance of Spok2. In all cases, these strains should not have been isolated from four-spored asci, indicating that either methodological issues occurred or that spore killing may still produce four-spored asci, but where the spores which should be absent are instead inviable.
Data availability
The full CDS sequence and UTRs of Spok3, Spok4, and SpokΨ1 (strain Wa87+) were deposited in NCBI GenBank under the accession numbers MK521588, MK521589, and MK521590, respectively. Raw sequencing reads were deposited on the NCBI SRA archive under the BioProject PRJNA523441. Final assemblies of samples with long-read data and alignment files (in fasta and nexus format) used to produce the main figures are available in the Dryad Digital Repository (https://doi.org/10.5061/dryad.vm1192g). Scripts and snakemake pipelines can be found at https://github.com/johannessonlab/SpokPaper (copy archived at https://github.com/elifesciences-publications/SpokPaper).
-
Dryad Digital RepositoryData from: Combinations of Spok genes create multiple meiotic drivers in Podospora.https://doi.org/10.5061/dryad.vm1192g
-
NCBI GenBankID MK521588. Full CDS sequence and UTRs of Spok3, Spok4, and Spok1 (strain Wa87+).
-
NCBI GenBankID MK521589. Full CDS sequence and UTRs of Spok3, Spok4, and Spok1 (strain Wa87+).
-
NCBI GenBankID MK521590. Full CDS sequence and UTRs of Spok3, Spok4, and Spok1 (strain Wa87+).
-
NCBI Sequence Read ArchiveID PRJNA523441. Raw sequencing reads.
-
European Nucleotide ArchiveID GCA_000226545.1. Podan2.
-
European Nucleotide ArchiveID GCA_900290415.1. PODCO.
References
-
Learning generative models for protein fold familiesProteins: Structure, Function, and Bioinformatics 79:1061–1078.https://doi.org/10.1002/prot.22934
-
SPAdes: a new genome assembly algorithm and its applications to single-cell sequencingJournal of Computational Biology 19:455–477.https://doi.org/10.1089/cmb.2012.0021
-
Natural variation of heterokaryon incompatibility gene het-c in podospora anserina reveals diversifying selectionMolecular Biology and Evolution 31:962–974.https://doi.org/10.1093/molbev/msu047
-
Trimmomatic: a flexible trimmer for illumina sequence dataBioinformatics 30:2114–2120.https://doi.org/10.1093/bioinformatics/btu170
-
Genetic villains: killer meiotic driversTrends in Genetics 34:424–433.https://doi.org/10.1016/j.tig.2018.02.003
-
ConferenceNeighborNet: an agglomerative method for the construction of planar phylogenetic networksInternational Workshop on Algorithms in Bioinformatics. pp. 375–391.https://doi.org/10.1007/3-540-45784-4_28
-
BookGenes in Conflict: The Biology of Selfish Genetic ElementsHarvard University Press.
-
BLAST+: architecture and applicationsBMC Bioinformatics 10:421.https://doi.org/10.1186/1471-2105-10-421
-
Genome annotation and curation using MAKER and MAKER-PCurrent Protocols in Bioinformatics 48:4.11.1–4.11.4.https://doi.org/10.1002/0471250953.bi0411s48
-
BookSeqinR 1.0-2: a contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysisIn: Bastolla U, Porto M, Roman H. E, Vendruscolo M, editors. Structural Approaches to Sequence Evolution. Heidelberg: Springer, Berlin. pp. 207–232.
-
The variant call format and VCFtoolsBioinformatics 27:2156–2158.https://doi.org/10.1093/bioinformatics/btr330
-
BCFtools/csq: haplotype-aware variant consequencesBioinformatics 33:2037–2039.https://doi.org/10.1093/bioinformatics/btx100
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Killer meiotic drive and dynamic evolution of the wtf gene familyMolecular Biology and Evolution 36:1201–1214.https://doi.org/10.1093/molbev/msz052
-
SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree buildingMolecular Biology and Evolution 27:221–224.https://doi.org/10.1093/molbev/msp259
-
Genome quality control: rip (repeat-induced point mutation) comes to podosporaMolecular Microbiology 40:586–595.https://doi.org/10.1046/j.1365-2958.2001.02367.x
-
Genes that bias mendelian segregationPLOS Genetics 10:e1004387.https://doi.org/10.1371/journal.pgen.1004387
-
The degenerate DNA transposon pat and repeat-induced point mutation (RIP) in podospora anserinaMolecular and General Genetics MGG 263:1061–1069.https://doi.org/10.1007/s004380050035
-
Genetic analysis of spore killing in the filamentous ascomycete podospora anserinaFungal Genetics and Biology 41:1088–1098.https://doi.org/10.1016/j.fgb.2004.08.008
-
Sex chromosome driveCold Spring Harbor Perspectives in Biology 7:a017616.https://doi.org/10.1101/cshperspect.a017616
-
A novel family of linear plasmids with homology to plasmid pAL2-1 of podospora anserinaMGG Molecular & General Genetics 246:638–647.https://doi.org/10.1007/BF00298971
-
Application of phylogenetic networks in evolutionary studiesMolecular Biology and Evolution 23:254–267.https://doi.org/10.1093/molbev/msj030
-
PrDOS: prediction of disordered protein regions from amino acid sequenceNucleic Acids Research 35:W460–W464.https://doi.org/10.1093/nar/gkm363
-
Snakemake-a scalable bioinformatics workflow engineBioinformatics 34:2520–2522.https://doi.org/10.1093/bioinformatics/bts480
-
Vcfr: a package to manipulate and visualize variant call format data in RMolecular Ecology Resources 17:44–53.https://doi.org/10.1111/1755-0998.12549
-
Circos: an information aesthetic for comparative genomicsGenome Research 19:1639–1645.https://doi.org/10.1101/gr.092759.109
-
Evidence for Recurrent Paralogous Gene Conversion and Exceptional Allelic Divergence in the Attacin Genes of Drosophila MelanogasterGenetics 159:659–671.
-
Minimap2: pairwise alignment for nucleotide sequencesBioinformatics 34:3094–3100.https://doi.org/10.1093/bioinformatics/bty191
-
The ecology and evolutionary dynamics of meiotic driveTrends in Ecology & Evolution 31:315–326.https://doi.org/10.1016/j.tree.2016.02.001
-
Gene identification in novel eukaryotic genomes by self-training algorithmNucleic Acids Research 33:6494–6506.https://doi.org/10.1093/nar/gki937
-
tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequenceNucleic Acids Research 25:955–964.https://doi.org/10.1093/nar/25.5.955
-
Segregation distortersAnnual Review of Genetics 25:511–581.https://doi.org/10.1146/annurev.ge.25.120191.002455
-
ConferenceProtein contact prediction by integrating joint evolutionary coupling analysis and supervised learningInternational Conference on Research in Computational Molecular Biology. pp. 218–221.https://doi.org/10.1007/978-3-319-16706-0_21
-
Icarus: visualizer for de novo assembly evaluationBioinformatics 32:3321–3323.https://doi.org/10.1093/bioinformatics/btw379
-
Challenges in homology search: hmmer3 and convergent evolution of coiled-coil regionsNucleic Acids Research 41:e121.https://doi.org/10.1093/nar/gkt263
-
IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogeniesMolecular Biology and Evolution 32:268–274.https://doi.org/10.1093/molbev/msu300
-
Genetic diversity of human pathogenic members of the fusarium oxysporum complex inferred from multilocus DNA sequence data and amplified fragment length polymorphism analyses: evidence for the recent dispersion of a geographically widespread clonal lineage and nosocomial originJournal of Clinical Microbiology 42:5109–5120.https://doi.org/10.1128/JCM.42.11.5109-5120.2004
-
[Mode of action of the genes responsible for abortion of certain products of meiosis in the Ascomycete, Podospora anserina]Comptes Rendus Hebdomadaires des Séances de l'Académie des Sciences 264:2300–2303.
-
MACSE v2: toolkit for the alignment of coding sequences accounting for frameshifts and stop codonsMolecular Biology and Evolution 35:2582–2584.https://doi.org/10.1093/molbev/msy159
-
The enemies within: intergenomic conflict, interlocus contest evolution (ICE), and the intraspecific red queenBehavioral Ecology and Sociobiology 41:1–10.https://doi.org/10.1007/s002650050357
-
Les phénomènes de barrage chez podospora anserina I. analyse génétique des barrages entre souches S et SRevue de Cytologie et de Cytophysiologie Vegetales 13:51–92.
-
Contribution à l’étude génétique d’un ascomycète tétrasporé: Podospora anserinaRhem Rv Cytol Biol Veg 11:201–304.
-
Meiotic drive in natural populations of Drosophila melanogaster. I. the Cytogenetic Basis of Segregation-DistortionGenetics 44:233–250.
-
Meiotic drive as an evolutionary forceThe American Naturalist 91:105–110.https://doi.org/10.1086/281969
-
Prospects for circumventing aminoglycoside kinase mediated antibiotic resistanceFrontiers in Cellular and Infection Microbiology 3:22.https://doi.org/10.3389/fcimb.2013.00022
-
BookPodospora anserina: from laboratory to biotechnologyIn: Horwitz BA, Mukherjee PA, Mukherjee M, Kubicek CP, editors. Genomics of Soil-and Plant-Associated Fungi Springer. Springer. pp. 283–309.https://doi.org/10.1007/978-3-642-39339-6_12
-
A gene graveyard in the genome of the fungus podospora comataMolecular Genetics and Genomics 294:177–190.https://doi.org/10.1007/s00438-018-1497-3
-
A RecB-family nuclease motif in the type I restriction endonuclease EcoR124INucleic Acids Research 36:3939–3949.https://doi.org/10.1093/nar/gkn333
-
Gene prediction with a hidden markov model and a new intron submodelBioinformatics 19 Suppl 2:ii215–ii225.https://doi.org/10.1093/bioinformatics/btg1080
-
Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamilyNucleic Acids Research 40:7016–7045.https://doi.org/10.1093/nar/gks382
-
Geographic distribution of neurospora spore killer strains and strains resistant to killingFungal Genetics and Biology 32:93–104.https://doi.org/10.1006/fgbi.2001.1253
-
Spore killer, a chromosomal factor in neurospora that kills meiotic products not containing itGenetics 93:587–606.
-
Meiotic Drive in Neurospora and Other FungiThe American Naturalist 137:416–429.https://doi.org/10.1086/285174
-
From FastQ data to High-Confidence variant calls: the genome analysis toolkit best practices pipelineIn Current Protocols in Bioinformatics pp. 11.10.1–11.1011.https://doi.org/10.1002/0471250953.bi1110s43
-
The dynamics of pAL2-1 homologous linear plasmids in podospora anserinaMolecular and General Genetics MGG 258:521–529.https://doi.org/10.1007/s004380050763
-
Spore-Killing Meiotic Drive Factors in a Natural Population of the Fungus Podospora anserinaGenetics 156:593–605.
-
Spore killing in the fungus Podospora anserina: a connection between meiotic drive and vegetative incompatibility?Genetica 117:59–65.
-
BUSCO applications from quality assessments to gene prediction and phylogenomicsMolecular Biology and Evolution.https://doi.org/10.1093/molbev/msx319
-
Selfish genetic elementsTrends in Ecology & Evolution 3:297–302.https://doi.org/10.1016/0169-5347(88)90105-X
-
Selfish genetic elements, genetic conflict, and evolutionary innovationPNAS 108 Suppl 2:10863–10870.https://doi.org/10.1073/pnas.1102343108
-
A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its coreJournal of Molecular Biology 430:2237–2243.https://doi.org/10.1016/j.jmb.2017.12.007
Article and author information
Author details
S Lorena Ament-Velásquez

Funding
H2020 European Research Council (ERC-2014-CoG)
- Hanna Johannesson
Swedish Research Council (Spore killer genomics)
- Hanna Johannesson
Lars Hierta Memorial Foundation (Podospora genomics)
- S Lorena Ament-Velásquez
Royal Physiographic Society in Lund (Podospora genomics)
- S Lorena Ament-Velásquez
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We would like to thank Magdalena Grudzinska-Sterno for valuable assistance with DNA and RNA extractions as well as library preparations. We are also thankful to Ola Wallerman for assistance with MinION Oxford Nanopore sequencing. We thank Mathieu Paoletti for discussion and access to RNA-seq pilot data at early stages of the project. We acknowledge the support of the National Genomics Infrastructure (NGI)/Uppsala Genome Center for assistance with massive parallel sequencing. The computations were performed on resources provided by SNIC through the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under Project SNIC 2017/1-567. This study was founded by a European Research Council grant under the program H2020, ERC-2014-CoG, project 648143 (SpoKiGen), with additional funding from The Swedish Research Council (VR) (to HJ), and from the Lars Hierta Memorial Foundation and The Nilsson-Ehle Endowments of the Royal Physiographic Society of Lund (to SLAV).
Version history
- Received: February 27, 2019
- Accepted: June 9, 2019
- Version of Record published: July 26, 2019 (version 1)
Copyright
© 2019, Vogan et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,146
- views
-
- 213
- downloads
-
- 58
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Combinations of Spok genes create multiple meiotic drivers in Podospora
eLife 8:e46454.
https://doi.org/10.7554/eLife.46454
Further reading
-
- Developmental Biology
- Evolutionary Biology
Despite rapid evolution across eutherian mammals, the X-linked MIR-506 family miRNAs are located in a region flanked by two highly conserved protein-coding genes (SLITRK2 and FMR1) on the X chromosome. Intriguingly, these miRNAs are predominantly expressed in the testis, suggesting a potential role in spermatogenesis and male fertility. Here, we report that the X-linked MIR-506 family miRNAs were derived from the MER91C DNA transposons. Selective inactivation of individual miRNAs or clusters caused no discernible defects, but simultaneous ablation of five clusters containing 19 members of the MIR-506 family led to reduced male fertility in mice. Despite normal sperm counts, motility, and morphology, the KO sperm were less competitive than wild-type sperm when subjected to a polyandrous mating scheme. Transcriptomic and bioinformatic analyses revealed that these X-linked MIR-506 family miRNAs, in addition to targeting a set of conserved genes, have more targets that are critical for spermatogenesis and embryonic development during evolution. Our data suggest that the MIR-506 family miRNAs function to enhance sperm competitiveness and reproductive fitness of the male by finetuning gene expression during spermatogenesis.
-
- Evolutionary Biology
- Immunology and Inflammation
CD4+ T cell activation is driven by five-module receptor complexes. The T cell receptor (TCR) is the receptor module that binds composite surfaces of peptide antigens embedded within MHCII molecules (pMHCII). It associates with three signaling modules (CD3γε, CD3δε, and CD3ζζ) to form TCR-CD3 complexes. CD4 is the coreceptor module. It reciprocally associates with TCR-CD3-pMHCII assemblies on the outside of a CD4+ T cells and with the Src kinase, LCK, on the inside. Previously, we reported that the CD4 transmembrane GGXXG and cytoplasmic juxtamembrane (C/F)CV+C motifs found in eutherian (placental mammal) CD4 have constituent residues that evolved under purifying selection (Lee et al., 2022). Expressing mutants of these motifs together in T cell hybridomas increased CD4-LCK association but reduced CD3ζ, ZAP70, and PLCγ1 phosphorylation levels, as well as IL-2 production, in response to agonist pMHCII. Because these mutants preferentially localized CD4-LCK pairs to non-raft membrane fractions, one explanation for our results was that they impaired proximal signaling by sequestering LCK away from TCR-CD3. An alternative hypothesis is that the mutations directly impacted signaling because the motifs normally play an LCK-independent role in signaling. The goal of this study was to discriminate between these possibilities. Using T cell hybridomas, our results indicate that: intracellular CD4-LCK interactions are not necessary for pMHCII-specific signal initiation; the GGXXG and (C/F)CV+C motifs are key determinants of CD4-mediated pMHCII-specific signal amplification; the GGXXG and (C/F)CV+C motifs exert their functions independently of direct CD4-LCK association. These data provide a mechanistic explanation for why residues within these motifs are under purifying selection in jawed vertebrates. The results are also important to consider for biomimetic engineering of synthetic receptors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}