Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. PfPGI-Correlated Mutation Analysis
2.2. Protein and Synthetic Gene Library
2.3. PfPGI Mutant Library
2.4. PfPGI Expression and Purification
2.5. PfPGI Activity Assay
2.6. PfPGI Crystallization
2.7. EPR Spectroscopy
2.8. Protein Data Bank Accession Codes
2.9. Protein Structure Preparation and Molecular Dynamics Simulations
2.10. AQUA-DUCT Analysis
3. Results and Discussion
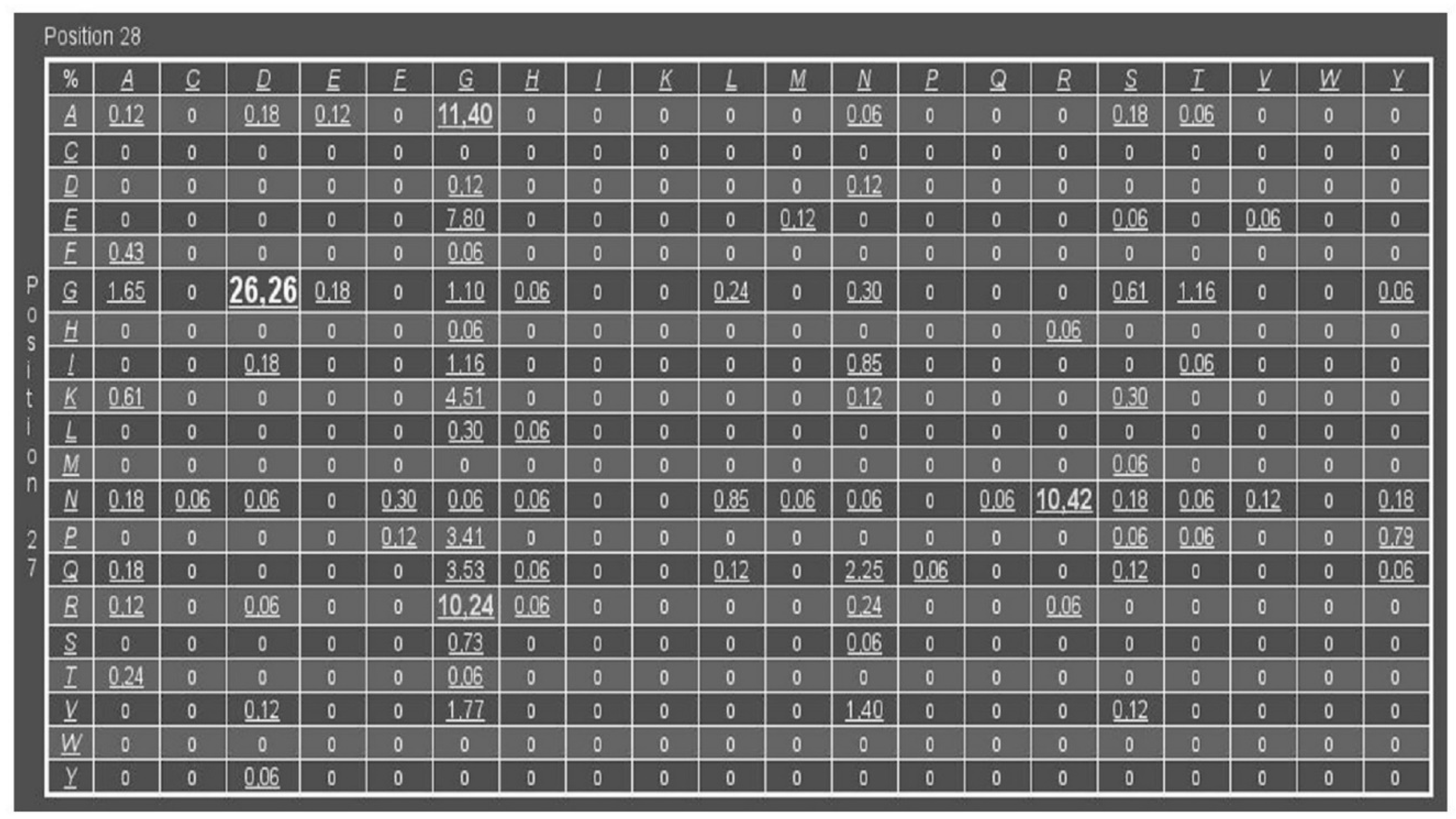
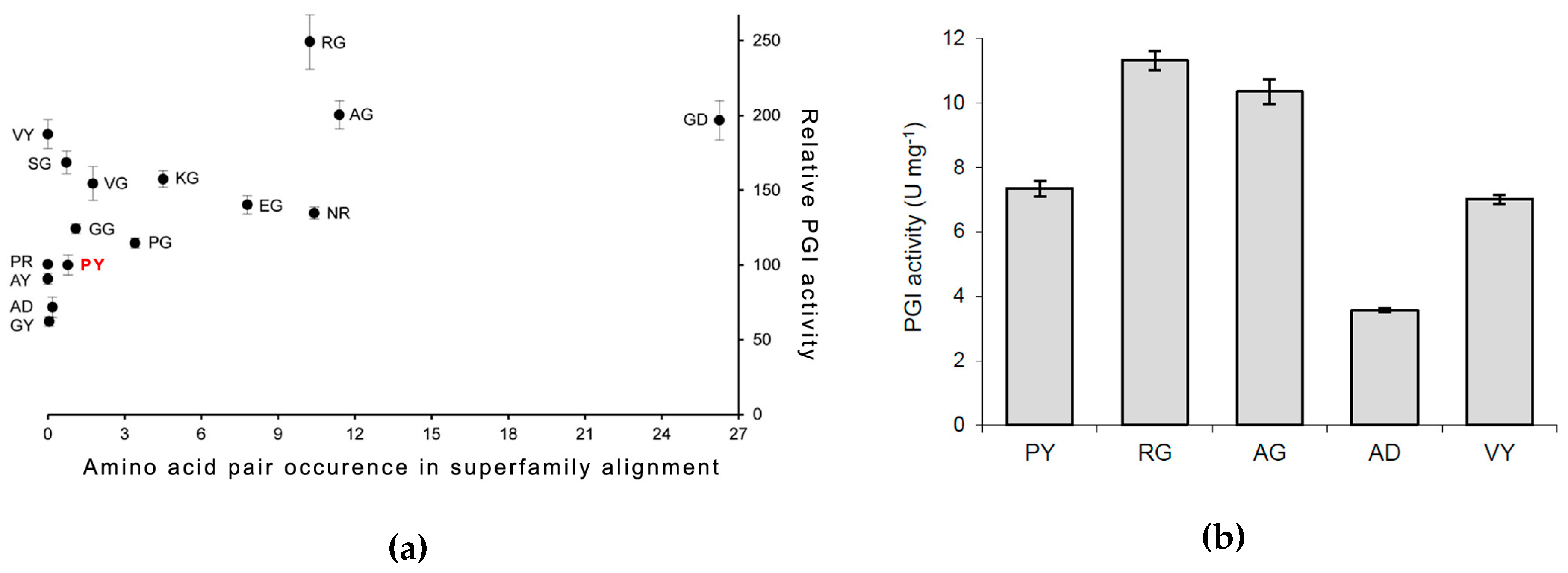
3.1. DM Analysis of Cupins and Comulator Predictions
3.2. PfPGI Mutant Activity Levels
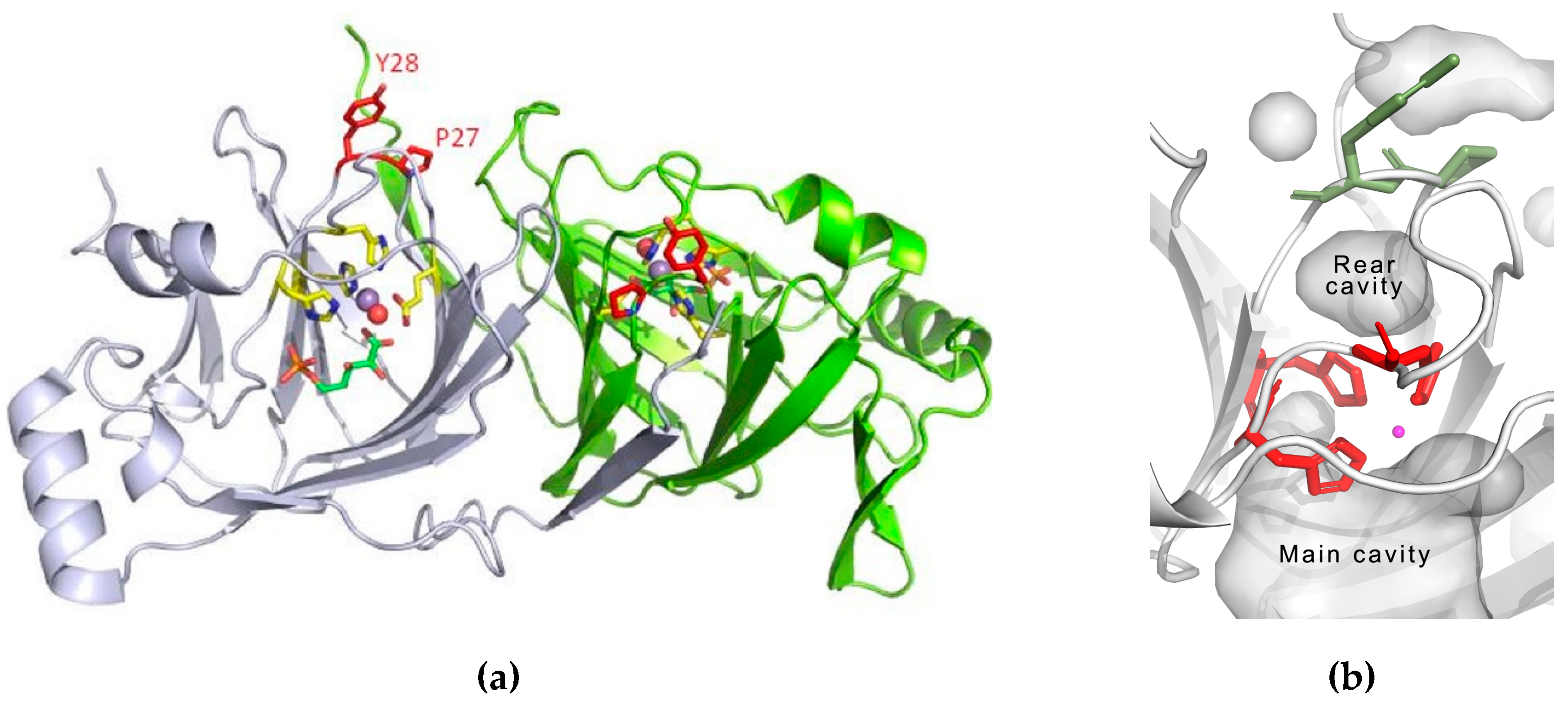
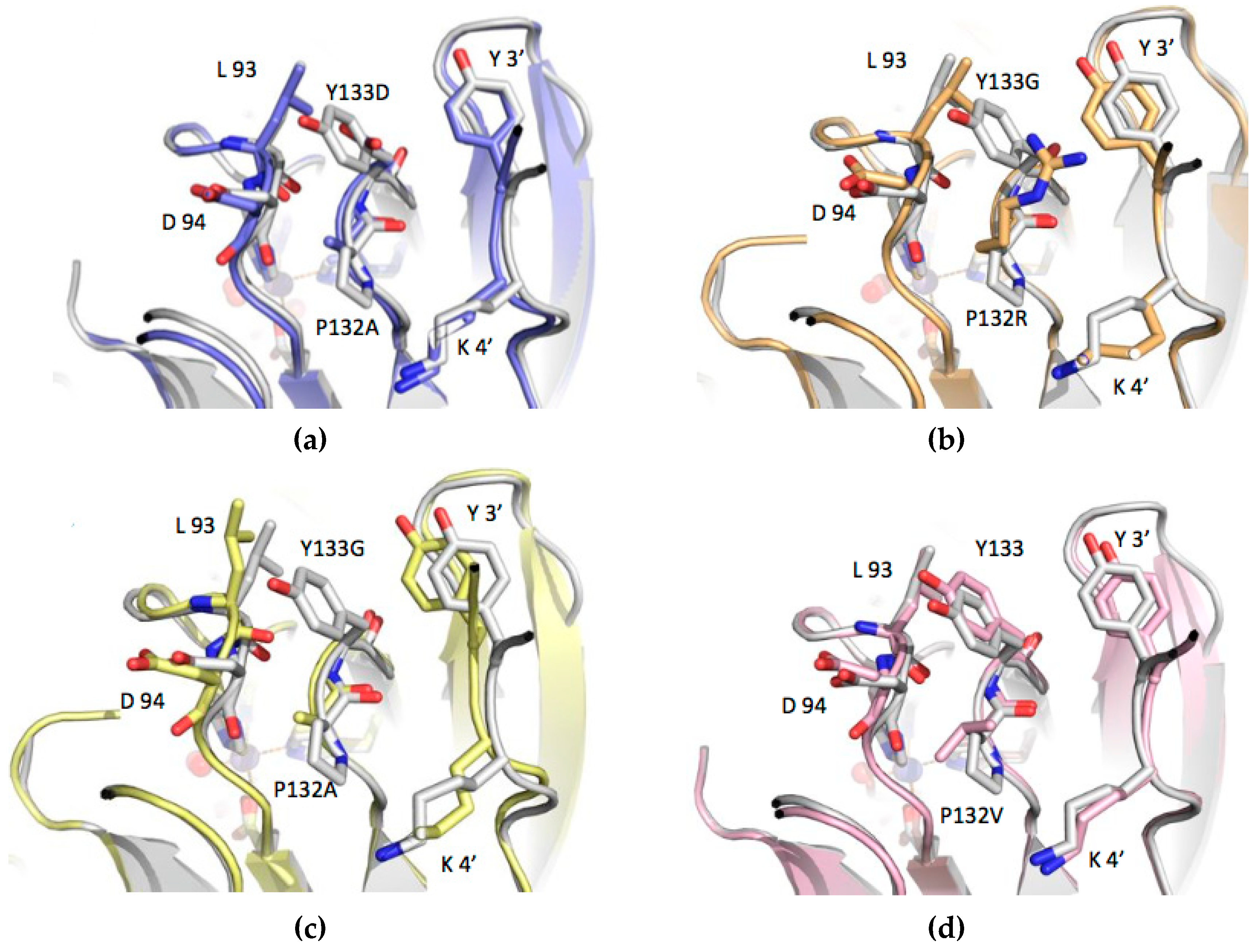
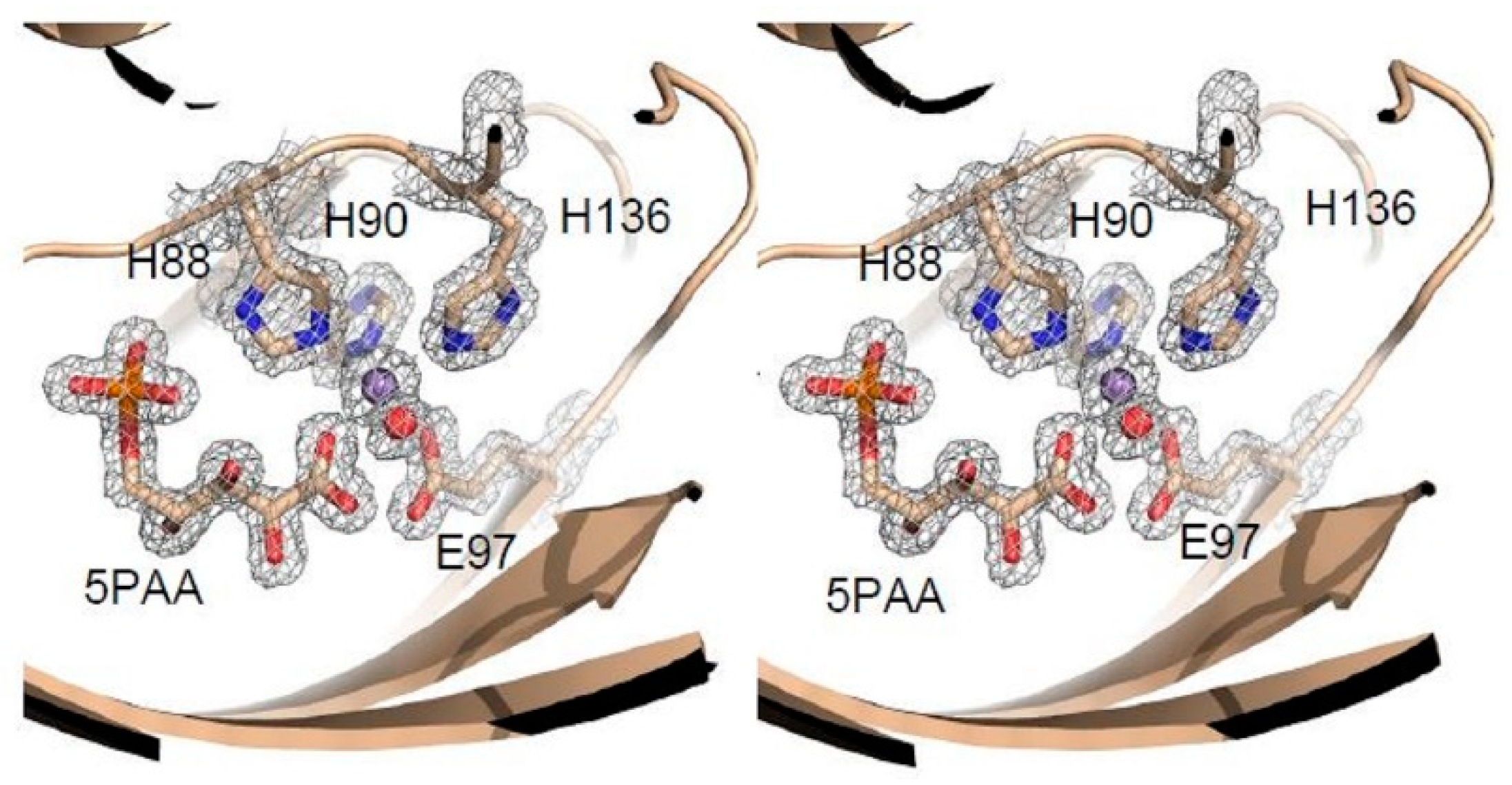
3.3. Structure Analysis of the Mutation-Carrying Loop
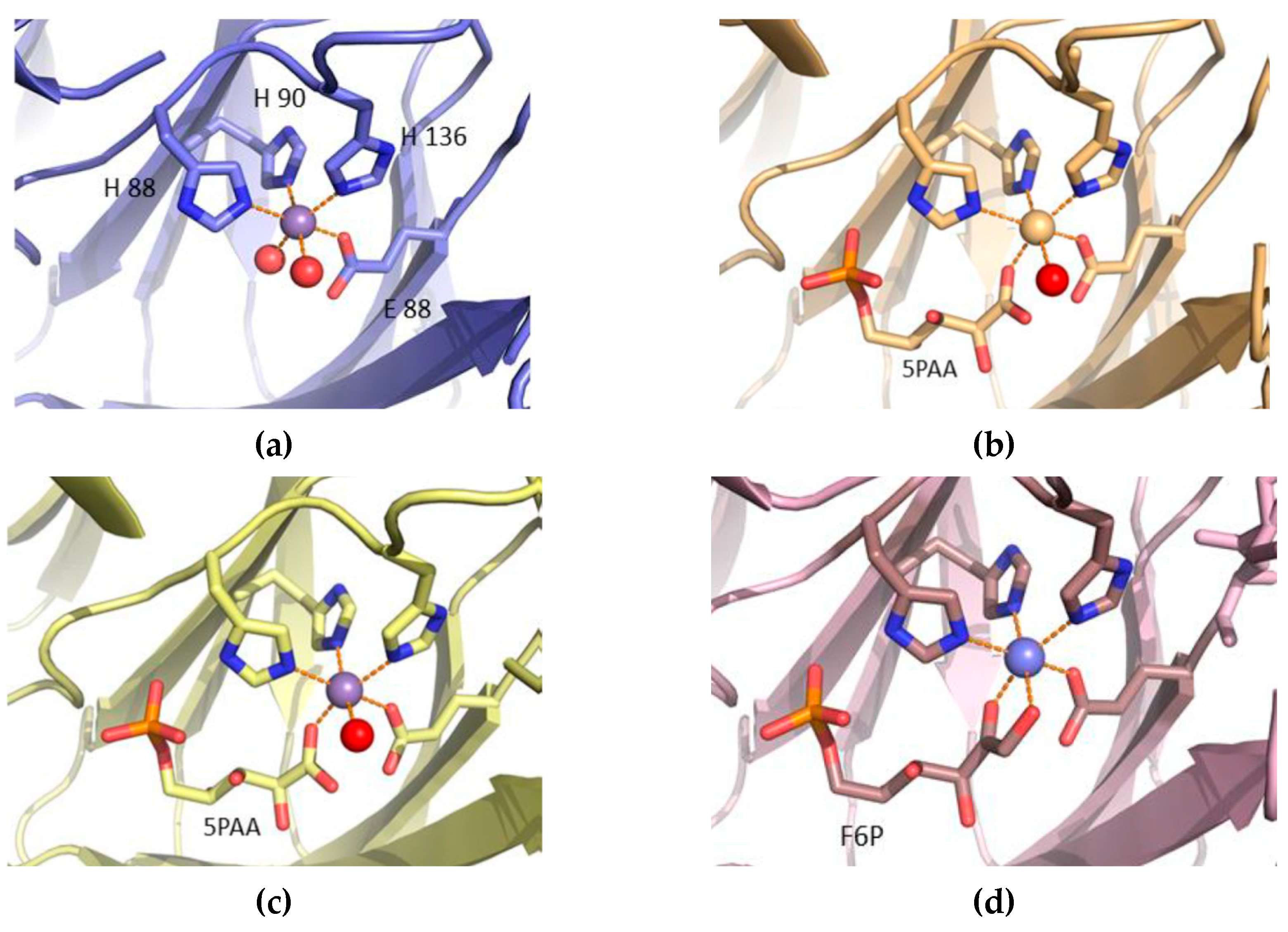
3.4. Structure Analysis of the Manganese Coordination
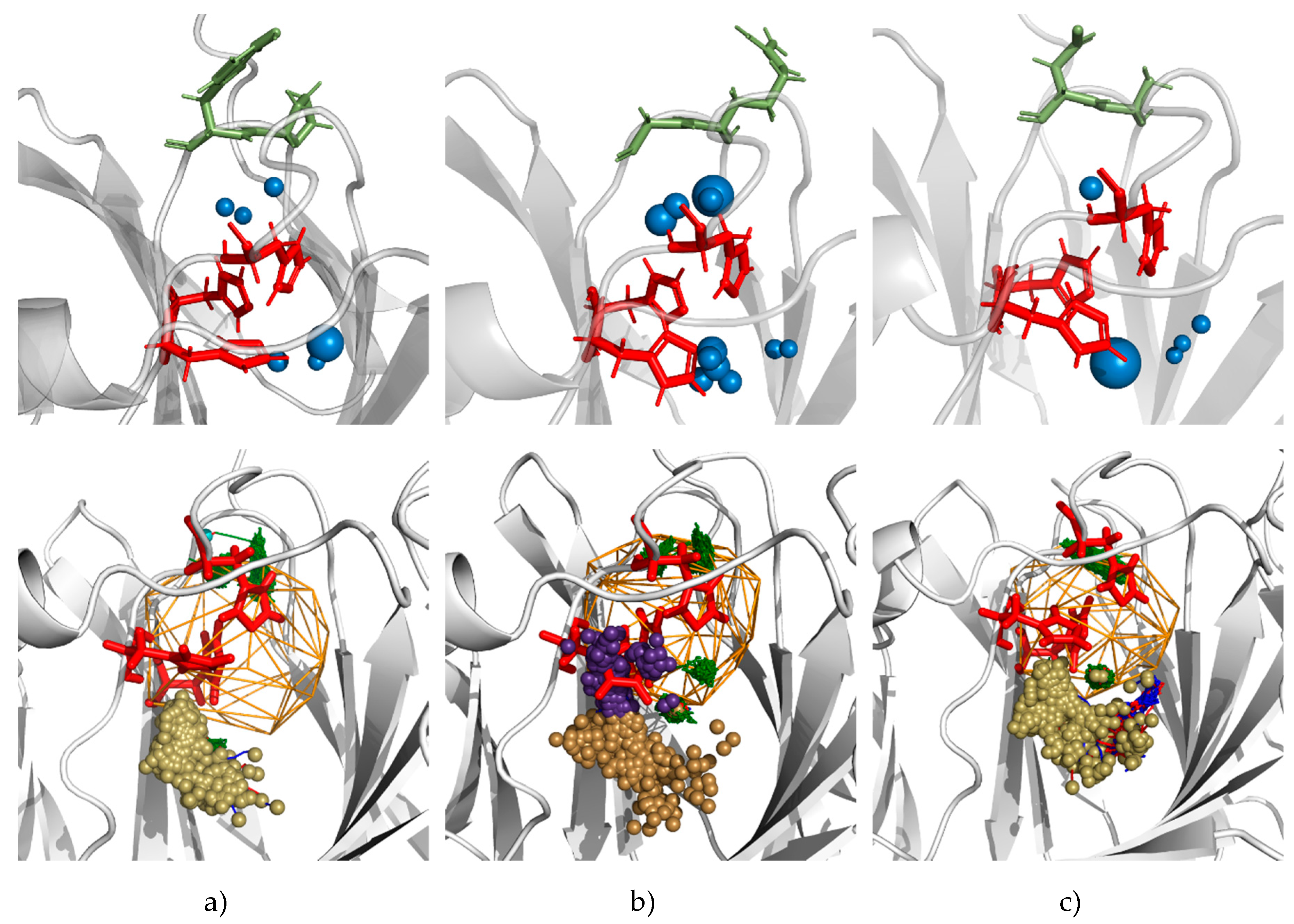
3.5. Molecular Dynamics Calculations and Water Access Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bendl, J.; Stourac, J.; Sebestova, E.; Vavra, O.; Musil, M.; Brezovsky, J.; Damborsky, J. HotSpot Wizard 2.0: Automated design of site-specific mutations and smart libraries in protein engineering. Nucleic Acids Res. 2016, 44, W479–W487. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef]
- Brunner, L.; Hoffmeister, H.E.; Koncz, J.; Stapenhorst, K. False diagnosis in constrictive pericarditis. Survey of clinical findings in 27 cases. Med. Klin. 1966, 61, 1283–1286. [Google Scholar] [PubMed]
- Fesko, K.; Suplatov, D.; Švedas, V. Bioinformatic analysis of the fold type I PLP-dependent enzymes reveals determinants of reaction specificity in l-threonine aldolase from Aeromonas jandaei. FEBS Open Bio 2018, 8, 1013–1028. [Google Scholar] [CrossRef]
- Lane, M.D.; Seelig, B. Advances in the directed evolution of proteins. Curr. Opin. Chem. Biol. 2014, 22, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Lutz, S. Beyond directed evolution—Semi-rational protein engineering and design. Curr. Opin. Biotechnol. 2010, 21, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, R.; Kim, J.Y.; Hong, E.Y.; Lee, S.; Seo, J.; Kim, B. RiSLnet: Rapid identification of smart mutant libraries using protein structure network. Application to thermal stability enhancement. Biotechnol. Bioeng. 2018. [Google Scholar] [CrossRef]
- Genz, M.; Melse, O.; Schmidt, S.; Vickers, C.; Dörr, M.; van den Bergh, T.; Joosten, H.J.; Bornscheuer, U.T. Engineering the Amine Transaminase from Vibrio fluvialis towards Branched-Chain Substrates. ChemCatChem 2016, 8, 3199–3202. [Google Scholar] [CrossRef]
- Moore, J.C.; Rodriguez-Granillo, A.; Crespo, A.; Govindarajan, S.; Welch, M.; Hiraga, K.; Lexa, K.; Marshall, N.; Truppo, M.D. “Site and Mutation”-Specific Predictions Enable Minimal Directed Evolution Libraries. ACS Synth Biol. 2018, 7, 1730–1741. [Google Scholar] [CrossRef]
- Nobili, A.; Gall, M.G.; Pavlidis, I.V.; Thompson, M.L.; Schmidt, M.; Bornscheuer, U.T. Use of ‘small but smart’ libraries to enhance the enantioselectivity of an esterase from Bacillus stearothermophilus towards tetrahydrofuran-3-yl acetate. FEBS J. 2013, 280, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, R.K.; Joosten, H.-J.; van Berkel, W.J.H.; Leferink, N.G.H.; Rooijen, E.; Ittmann, E.; van Zimmeren, F.; Jochens, H.; Bornscheuer, U.; Vriend, G.; et al. 3DM: Systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins Struct. Funct. Bioinform. 2010. [Google Scholar] [CrossRef] [PubMed]
- Kundrotas, P.J.; Alexov, E.G. Predicting residue contacts using pragmatic correlated mutations method: Reducing the false positives. BMC Bioinform. 2006, 7, 503. [Google Scholar] [CrossRef] [PubMed]
- Mintseris, J.; Weng, Z. Structure, function, and evolution of transient and obligate protein-protein interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 10930–10935. [Google Scholar] [CrossRef] [PubMed]
- Pazos, F.; Helmer-Citterich, M.; Ausiello, G.; Valencia, A. Correlated mutations contain information about protein-protein interaction. J. Mol. Biol. 1997, 271, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kowarsch, A.; Fuchs, A.; Frishman, D.; Pagel, P. Correlated Mutations: A Hallmark of Phenotypic Amino Acid Substitutions. PLoS Comput. Biol. 2010, 6, e1000923. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Martin, L.C.; Wahl, L.M.; Dunn, S.D. Mutual Information in Protein Multiple Sequence Alignments Reveals Two Classes of Coevolving Positions. Biochemistry 2005, 44, 7156–7165. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, T.; Tamo, G.; Nobili, A.; Tao, Y.; Tan, T.; Bornscheuer, U.T.; Kuipers, R.; de Jong, V.; Subramanian, K.; Schaap, P.; et al. CorNet: Assigning function to networks of co-evolving residues by automated literature mining. PLoS ONE 2017, 12, e0176427. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, J.M.; Khuri, S.; Gane, P.J. Microbial Relatives of the Seed Storage Proteins of Higher Plants: Conservation of Structure and Diversification of Function during Evolution of the Cupin Superfamily. Microbiol. Mol. Biol. Rev. 2000, 64, 153–179. [Google Scholar] [CrossRef] [Green Version]
- Dunwell, J.M.; Purvis, A.; Khuri, S. Cupins: The most functionally diverse protein superfamily? Phytochemistry 2004, 65, 7–17. [Google Scholar] [CrossRef]
- DiRuggiero, J.; Santangelo, N.; Nackerdien, Z.; Ravel, J.; Robb, F.T. Repair of extensive ionizing-radiation DNA damage at 95 degrees C in the hyperthermophilic archaeon Pyrococcus furiosus. J. Bacteriol. 1997, 179, 4643–4645. [Google Scholar] [CrossRef] [Green Version]
- Hansen, T.; Oehlmann, M.; Schonheit, P. Novel Type of Glucose-6-Phosphate Isomerase in the Hyperthermophilic Archaeon Pyrococcus furiosus. J. Bacteriol. 2001, 183, 3428–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhees, C.H.; Huynen, M.A.; Ward, D.E.; Schiltz, E.; de Vos, W.M.; van der Oost, J. The Phosphoglucose Isomerase from the Hyperthermophilic Archaeon Pyrococcus furiosus Is a Unique Glycolytic Enzyme That Belongs to the Cupin Superfamily. J. Biol. Chem. 2001, 276, 40926–40932. [Google Scholar] [CrossRef]
- Berrisford, J.M.; Akerboom, J.; Brouns, S.; Sedelnikova, S.E.; Turnbull, A.P.; van der Oost, J.; Salmon, L.; Hardré, R.; Murray, I.A.; Blackburn, G.M.; et al. The Structures of Inhibitor Complexes of Pyrococcus furiosus Phosphoglucose Isomerase Provide Insights into Substrate Binding and Catalysis. J. Mol. Biol. 2004, 343, 649–657. [Google Scholar] [CrossRef]
- Berrisford, J.M.; Akerboom, J.; Turnbull, A.P.; de Geus, D.; Sedelnikova, S.E.; Staton, I.; McLeod, C.; Verhees, C.; van der Oost, J.; Rice, D.; et al. Crystal Structure of Pyrococcus furiosus Phosphoglucose Isomerase. J. Biol. Chem. 2003, 278, 33290–33297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrisford, J.M.; Hounslow, A.M.; Akerboom, J.; Hagen, W.R.; Brouns, S.J.J.; van der Oost, J.; Murray, I.A.; Michael Blackburn, G.; Waltho, J.P.; Rice, D.W.; et al. Evidence Supporting a cis-enediol-based Mechanism for Pyrococcus furiosus Phosphoglucose Isomerase. J. Mol. Biol. 2006, 358, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Schlichting, B.; Felgendreher, M.; Schonheit, P. Cupin-Type Phosphoglucose Isomerases (Cupin-PGIs) Constitute a Novel Metal-Dependent PGI Family Representing a Convergent Line of PGI Evolution. J. Bacteriol. 2005, 187, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.; Xie, H.; Cao, Z.; Mo, Y. Combined Quantum Mechanics/Molecular Mechanics Study on the Reversible Isomerization of Glucose and Fructose Catalyzed by Pyrococcus furiosus Phosphoglucose Isomerase. J. Am. Chem. Soc. 2008, 130, 7022–7031. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Goodey, N.M. Catalytic Contributions from Remote Regions of Enzyme Structure. Chem. Rev. 2011, 111, 7595–7624. [Google Scholar] [CrossRef] [PubMed]
- Deniz, A.A. Enzymes can adapt to cold by wiggling regions far from their active site. Nature 2018, 558, 195–196. [Google Scholar] [CrossRef] [Green Version]
- Saavedra, H.G.; Wrabl, J.O.; Anderson, J.A.; Li, J.; Hilser, V.J. Dynamic allostery can drive cold adaptation in enzymes. Nature 2018, 558, 324–328. [Google Scholar] [CrossRef]
- Kuipers, R.K.P.; Joosten, H.-J.; Verwiel, E.; Paans, S.; Akerboom, J.; van der Oost, J.; Leferink, N.G.; van Berkel, W.J.; Vriend, G.; Schaap, P.J. Correlated mutation analyses on super-family alignments reveal functionally important residues. Proteins Struct. Funct. Bioinform. 2009, 76, 608–616. [Google Scholar] [CrossRef]
- Akerboom, J.; Turnbull, A.P.; Hargreaves, D.; Fisher, M.; de Geus, D.; Sedelnikova, S.E.; Berrisford, J.M.; Baker, P.J.; Verhees, C.H.; van der Oost, J.; et al. Purification, crystallization and preliminary crystallographic analysis of phosphoglucose isomerase from the hyperthermophilic archaeon Pyrococcus furiosus. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1822–1823. [Google Scholar] [CrossRef]
- Winter, G. Xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. Sect. D Biol. Crystallogr. 1997. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. AMBER14, University of California: San Francisco, CA, USA, 2014.
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for +2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magdziarz, T.; Mitusińska, K.; Gołdowska, S.; Płuciennik, A.; Stolarczyk, M.; Lugowska, M.; Góra, A. AQUA-DUCT: A ligands tracking tool. Bioinformatics 2017, 33, 2045–2046. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System, DeLano Scientific: Palo Alto, CA, USA, 2002.
- Mitusińska, K.; Magdziarz, T.; Bzówka, M.; Stańczak, A.; Gora, A. Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. Biomolecules 2018, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Dudev, T.; Lim, C. Monodentate versus Bidentate Carboxylate Binding in Magnesium and Calcium Proteins: What Are the Basic Principles? J. Phys. Chem. B 2004, 108, 4546–4557. [Google Scholar] [CrossRef]
- Hayden, J.A.; Brophy, M.B.; Cunden, L.S.; Nolan, E.M. High-Affinity Manganese Coordination by Human Calprotectin Is Calcium-Dependent and Requires the Histidine-Rich Site Formed at the Dimer Interface. J. Am. Chem. Soc. 2013, 135, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Azmat, R.; Naz, R.; Qamar, N.; Malik, I. Kinetics and mechanisms of oxidation of d-fructose and d-lactose by permanganate ion in acidic medium. Nat. Sci. 2012, 4, 466–478. [Google Scholar] [CrossRef]
- Csermely, P.; Palotai, R.; Nussinov, R. Induced fit, conformational selection and independent dynamic segments: An extended view of binding events. Trends Biochem. Sci. 2010, 35, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Teşileanu, T.; Colwell, L.J.; Leibler, S. Protein Sectors: Statistical Coupling Analysis versus Conservation. Wilke CO, editor. PLoS Comput. Biol. 2015, 11, e1004091. [Google Scholar] [CrossRef]
- Gora, A.; Brezovsky, J.; Damborsky, J. Gates of Enzymes. Chem. Rev. 2013, 113, 5871–5923. [Google Scholar] [CrossRef]
- Subramanian, K.; Góra, A.; Spruijt, R.; Mitusińska, K.; Suarez-Diez, M.; Martins dos Santos, V.; Schaap, P.J. Modulating D-amino acid oxidase (DAAO) substrate specificity through facilitated solvent access. PLoS ONE 2018, 13, e0198990. [Google Scholar] [CrossRef]
- Persson, F.; Halle, B. Transient access to the protein interior: Simulation versus NMR. J. Am. Chem. Soc. 2013, 135, 8735–8748. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramanian, K.; Mitusińska, K.; Raedts, J.; Almourfi, F.; Joosten, H.-J.; Hendriks, S.; Sedelnikova, S.E.; Kengen, S.W.M.; Hagen, W.R.; Góra, A.; et al. Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase. Biomolecules 2019, 9, 212. https://doi.org/10.3390/biom9060212
Subramanian K, Mitusińska K, Raedts J, Almourfi F, Joosten H-J, Hendriks S, Sedelnikova SE, Kengen SWM, Hagen WR, Góra A, et al. Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase. Biomolecules. 2019; 9(6):212. https://doi.org/10.3390/biom9060212
Chicago/Turabian StyleSubramanian, Kalyanasundaram, Karolina Mitusińska, John Raedts, Feras Almourfi, Henk-Jan Joosten, Sjon Hendriks, Svetlana E. Sedelnikova, Servé W. M. Kengen, Wilfred R. Hagen, Artur Góra, and et al. 2019. "Distant Non-Obvious Mutations Influence the Activity of a Hyperthermophilic Pyrococcus furiosus Phosphoglucose Isomerase" Biomolecules 9, no. 6: 212. https://doi.org/10.3390/biom9060212