Metaproteomic and 16S rRNA Gene Sequencing Analysis of the Infant Fecal Microbiome

 ,
,
Abstract
:1. Introduction
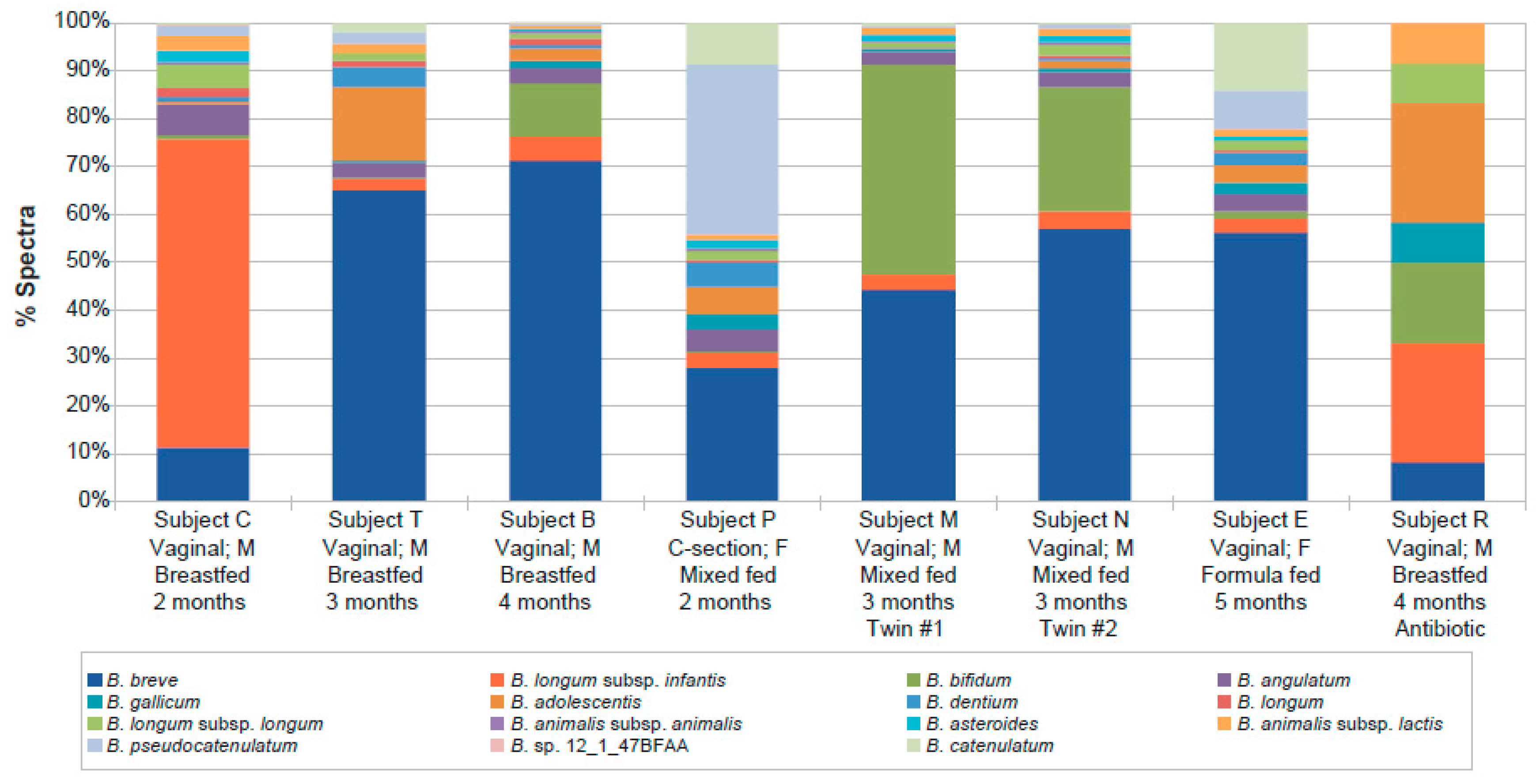
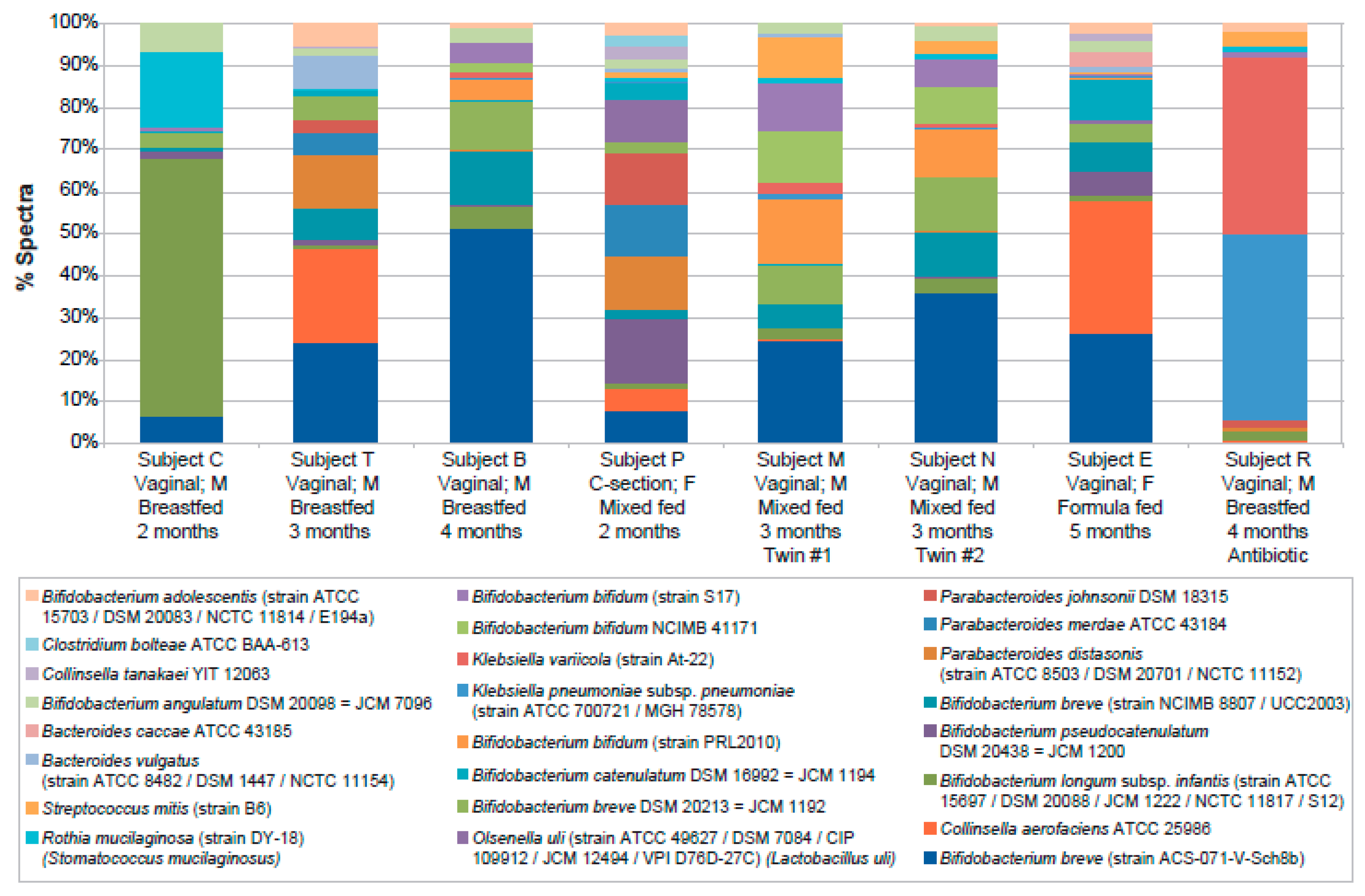
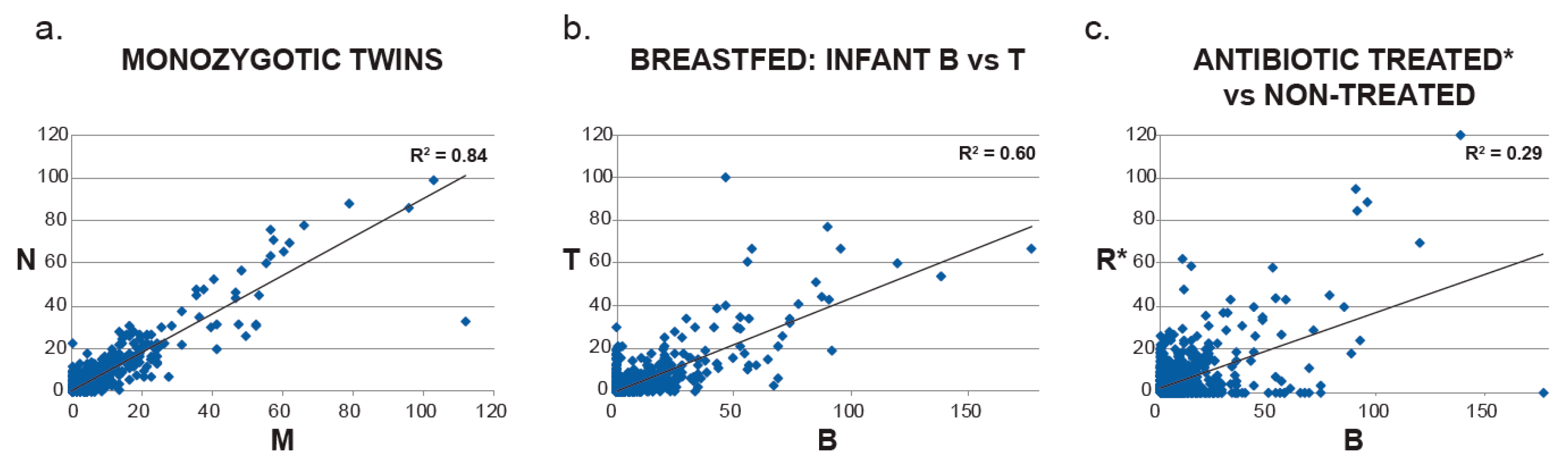
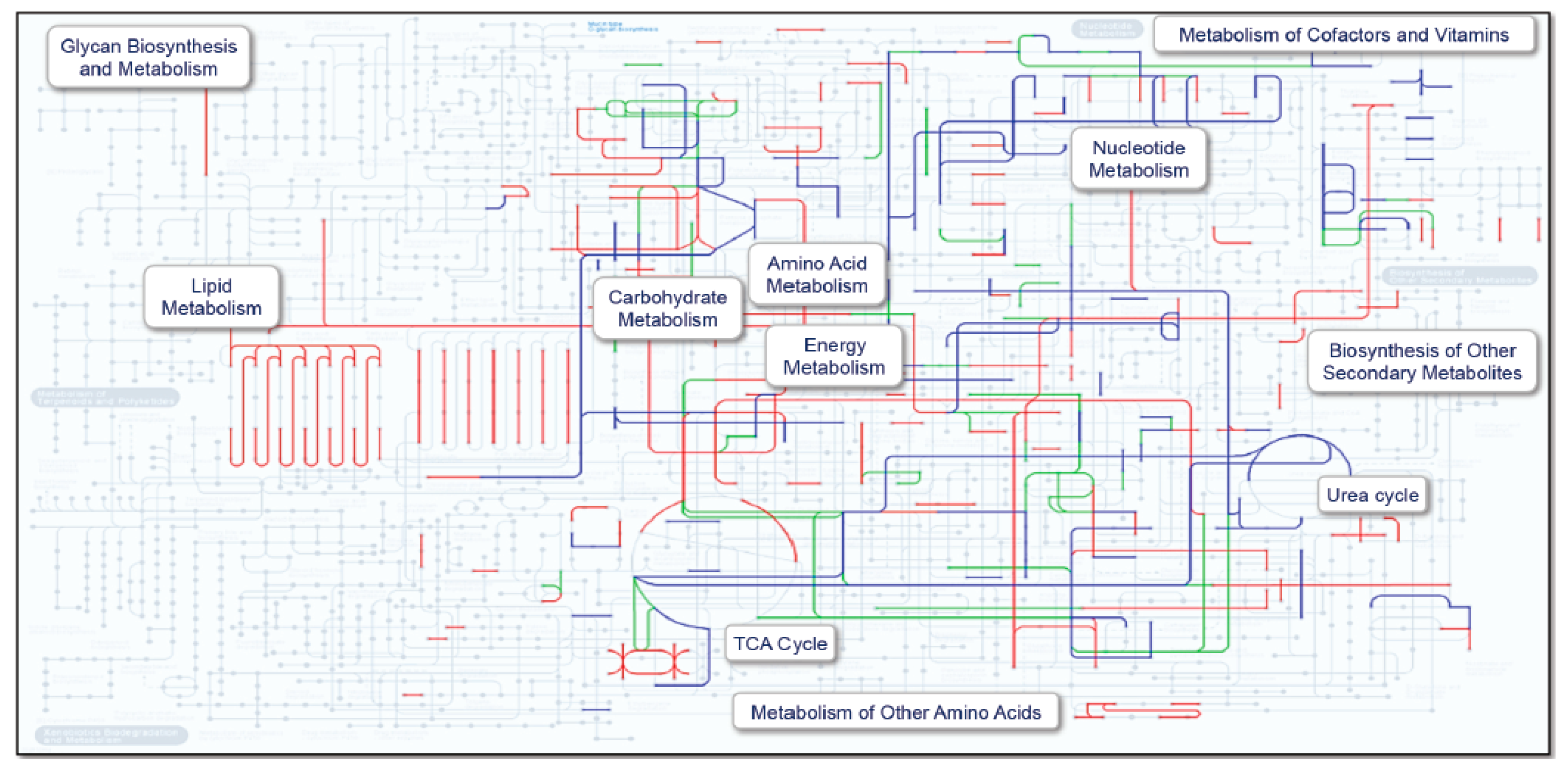
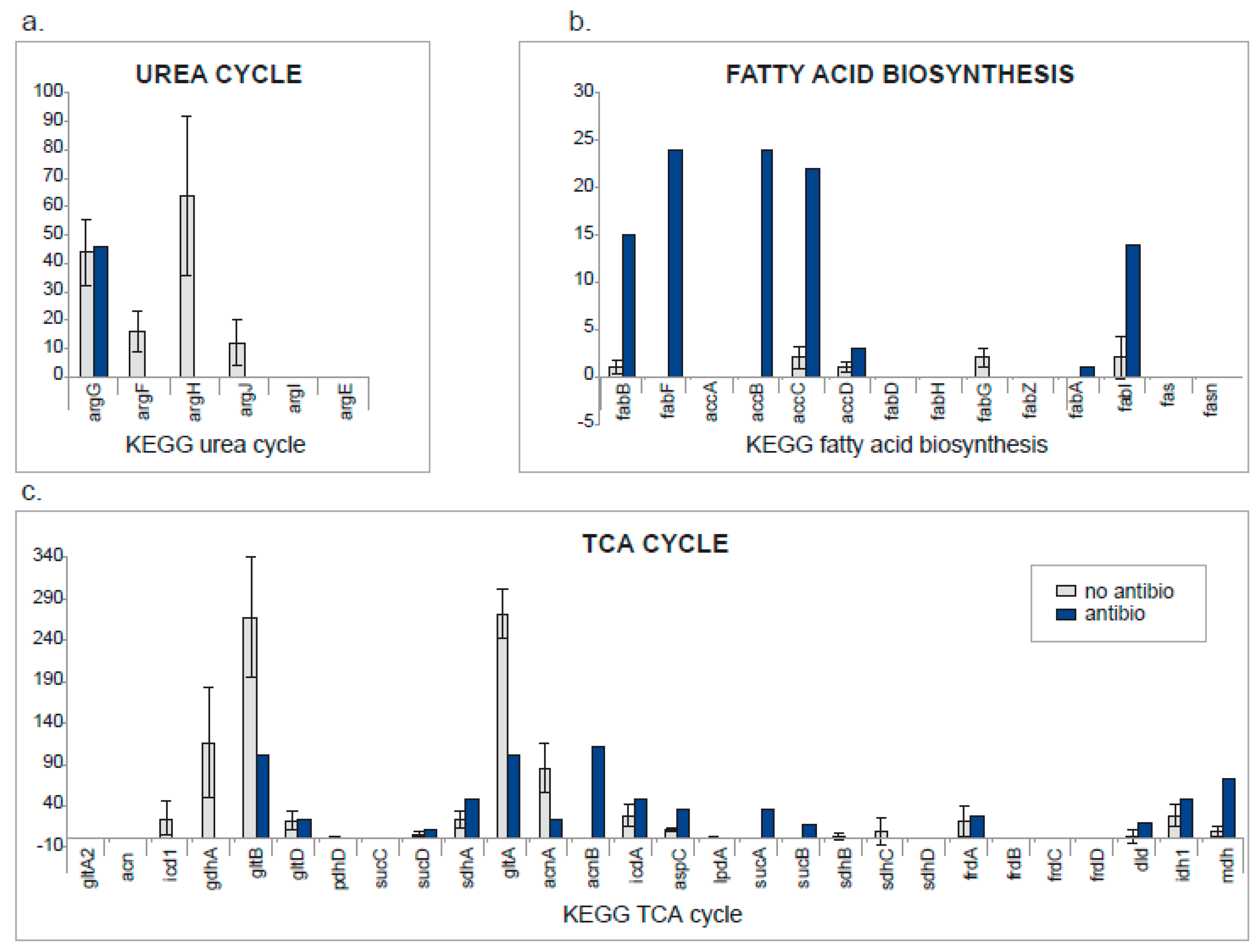
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. 16S rRNA Gene Sequencing and Bioinformatics
4.3. Proteomics Sample Preparation and Analysis
4.4. Availability of Data and Materials
4.5. Ethics Approval and Consent to Participate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| C-section | Caesarean-section |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| TCA | tricarboxylic acid |
| HMO | human milk oligosaccharides |
| SCXUPLC | strong cation exchangeultra performance liquid chromatography |
References
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyotylainen, T.; Hamalainen, A.M.; Peet, A.; Tillmann, V.; Poho, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, H.; Oozeer, R.; Knipping, K.; Belzer, C.; Knol, J. The first thousand days—Intestinal microbiology of early life: Establishing a symbiosis. Pediatr. Allergy Immunol. 2014, 25, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Chung, J.; Battaglia, T.; Henderson, N.; Jay, M.; Li, H.; DLieber, A.; Wu, F.; Perez-Perez, G.I.; Chen, Y.; et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016, 8, 343ra82. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hämäläinen, A.-M.; Härkönen, T.; Ryhänen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016, 8, 343ra81. [Google Scholar] [CrossRef] [PubMed]
- Korpela, K.; Salonen, A.; Virta, L.J.; Kekkonen, R.A.; de Vos, W.M. Association of Early-Life Antibiotic Use and Protective Effects of Breastfeeding: Role of the Intestinal Microbiota. JAMA Pediatr. 2016, 170, 750–757. [Google Scholar] [CrossRef]
- Langdon, A.; Crook, N.; Dantas, G. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med. 2016, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Chari, R.S.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L. Gut microbiota of healthy Canadian infants: Profiles by mode of delivery and infant diet at 4 months. Can. Med. Assoc. J. 2013, 185, 385–394. [Google Scholar] [CrossRef]
- Chen, J.; Cai, W.; Feng, Y. Development of intestinal bifidobacteria and lactobacilli in breast-fed neonates. Clin. Nutr. 2007, 26, 559–566. [Google Scholar] [CrossRef]
- Jost, T.; Lacroix, C.; Braegger, C.P.; Chassard, C. New Insights in Gut Microbiota Establishment in Healthy Breast Fed Neonates. PLoS ONE 2012, 7, e44595. [Google Scholar] [CrossRef] [Green Version]
- Roger, L.C.; Costabile, A.; Holland, D.T.; Hoyles, L.; McCartney, A.L. Examination of faecal Bifidobacterium populations in breast- and formula-fed infants during the first 18 months of life. Microbiology 2010, 156, 3329–3341. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Makino, H.; Cetinyurek Yavuz, A.; Ben-Amor, K.; Roelofs, M.; Ishikawa, E.; Kubota, H.; Swinkels, S.; Sakai, T.; Oishi, K.; et al. Early-Life Events, Including Mode of Delivery and Type of Feeding, Siblings and Gender, Shape the Developing Gut Microbiota. PLoS ONE 2016, 11, e0158498. [Google Scholar] [CrossRef]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef] [Green Version]
- Putignani, L.; Del Chierico, F.; Petrucca, A.; Vernocchi, P.; Dallapiccola, B. The human gut microbiota: A dynamic interplay with the host from birth to senescence settled during childhood. Pediatr. Res. 2014, 76, 2–10. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Bergström, A.; Skov, T.H.; Bahl, M.I.; Roager, H.M.; Christensen, L.B.; Ejlerskov, K.T.; Mølgaard, C.; Michaelsen, K.F.; Licht, T.R. Establishment of Intestinal Microbiota during Early Life: A Longitudinal, Explorative Study of a Large Cohort of Danish Infants. Appl. Environ. Microbiol. 2014, 80, 2889–2900. [Google Scholar] [CrossRef]
- Huda, M.N.; Lewis, Z.; Kalanetra, K.M.; Rashid, M.; Ahmad, S.M. Stool Microbiota and Vaccine Responses of Infants. Pediatrics 2014, 134. [Google Scholar] [CrossRef]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. System. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef] [Green Version]
- Sim, K.; Cox, M.J.; Wopereis, H.; Martin, R.; Knol, J.; Li, M.-S.; Cookson, W.O.C.M.; Moffatt, M.F.; Kroll, J.S. Improved Detection of Bifidobacteria with Optimised 16S rRNA-Gene Based Pyrosequencing. PLoS ONE 2012, 7, e32543. [Google Scholar] [CrossRef]
- Case, R.J.; Boucher, Y.; Dahllöf, I.; Holmström, C.; Doolittle, W.F.; Kjelleberg, S. Use of 16S rRNA and rpoB Genes as Molecular Markers for Microbial Ecology Studies. Appl. Environ. Microbiol. 2007, 73, 278–288. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S rRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. System. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Land, M.; Hauser, L.; Jun, S.-R.; Nookaew, I.; Leuze, M.R.; Ahn, T.-H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Function. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.B.; Highlander, S.K.; Anderson, E.L.; Li, W.; Dayrit, M.; Klitgord, N.; Fabani, M.M.; Seguritan, V.; Green, J.; Pride, D.T.; et al. Library preparation methodology can influence genomic and functional predictions in human microbiome research. Proc. Natl. Acad. Sci. USA 2015, 112, 14024–14029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, R.; Abnet, C.C.; White, O.; Knight, R.; Huttenhower, C. The microbiome quality control project: Baseline study design and future directions. Genome Biol. 2015, 16, 276. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Heintz-Buschart, A.; Bond, P. A decade of metaproteomics: Where we stand and what the future hold. Proteomics 2015, 15, 3409–3417. [Google Scholar] [CrossRef]
- Xiong, W.; Abraham, P.E.; Li, Z.; Pan, C.; Hettich, R.L. Microbial metaproteomics for characterizing the range of metabolic functions and activities of human gut microbiota. Proteomics 2015, 15, 3424–3438. [Google Scholar] [PubMed] [Green Version]
- Kolmeder, C.A.; de Vos, W.M. Metaproteomics of our microbiome -developing insight in function and activity in man and model systems. J. Proteom. 2014, 97, 3–16. [Google Scholar] [CrossRef]
- Abraham, P.; Adams, R.; Giannone, R.J.; Kalluri URanjan, P.; Erickson, B.; Shah, M.; Tuskan, G.A.; Hettich, R.L. Defining the boundaries and characterizing the landscape of functional genome expression in vascular tissues of Populus using shotgun proteomics. J. Proteome Res. 2011, 11, 449–460. [Google Scholar] [CrossRef]
- VerBerkmoes, N.C.; Denef, V.J.; Hettich, R.L.; Banfield, J.F. Systems biology: Functional analysis of natural microbial consortia using community proteomics. Nat. Rev. Microbiol. 2009, 7, 196–205. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, W.; Ning, Z.; Mayne, J.; Mack, D.; Stintzi, A.; Tian, R.; Figeys, D. Deep Metaproteomics Approach for the Study of Human Microbiomes. Anal. Chem. 2017, 89, 9407–9415. [Google Scholar] [CrossRef]
- Young, J.C.; Pan, C.; Adams, R.; Brooks, B.; Banfield, J.F.; Morowitz, M.J.; Hettich, R.L. Metaproteomics Reveals Functional Shifts in Microbial and Human Proteins During a Preterm Infant Gut Colonization Case. Proteomics 2015, 15, 3463–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaassens, E.S.; de Vos, W.M.; Vaughan, E.E. Metaproteomics Approach to Study the Functionality of the Microbiota in the Human Infant Gastrointestinal Tract. Appl. Environ. Microbiol. 2007, 73, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Giannone, R.J.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Development of an enhanced metaproteomic approach for deepening the microbiome characterization of the human infant gut. J. Proteome Res. 2015, 14, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Brown, C.T.; Morowitz, M.J.; Banfield, J.F.; Hettich, R.L. Genome-resolved metaproteomic characterization of preterm infant gut microbiota development reveals species-specific metabolic shifts and variabilities during early life. Microbiome 2017, 5, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwittink, R.D.; van Zoeren-Grobben, D.; Martin, R.; van Lingen, R.A.; Groot Jebbink, L.J.; Boeren, S.; Renes, I.B.; van Elburg, R.M.; Belzer, C.; Knol, J. Metaproteomics reveals functional differences in intestinal microbiota development of preterm infants. Mol. Cell Proteom. 2017, 16, 1610–1620. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Ruiz-Moyano, S.; Totten, S.M.; Garrido, D.A.; Smilowitz, J.T.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Variation in Consumption of Human Milk Oligosaccharides by Infant Gut-Associated Strains of Bifidobacterium breve. Appl. Environ. Microbiol. 2013, 79, 6040–6049. [Google Scholar] [CrossRef] [Green Version]
- Marcobal, A.; Barboza, M.; Sonnenburg, E.D.; Pudlo, N.; Martens, E.C.; Desai, P.; Lebrilla, C.B.; Weimer, B.C.; Mills, D.A.; German, J.B.; et al. Bacteroides in the Infant Gut Consume Milk Oligosaccharides via Mucus-Utilization Pathways. Cell Host Microbe 2011, 10, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef]
- Junick, J.; Blaut, M. Quantification of human fecal bifidobacterium species by use of quantitative real-time PCR analysis targeting the groEL gene. Appl. Environ. Microbiol. 2012, 78, 2613–2622. [Google Scholar] [CrossRef]
- Guaraldi, F.; Salvatori, G. Effect of Breast and Formula Feeding on Gut Microbiota Shaping in Newborns. Front. Cell. Infect. Microbiol. 2012, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.A.; German, J.B.; Lebrilla, C.B.; Mills, D.A. Bifidobacterium longum subspecies infantis: Champion colonizer of the infant gut. Pediatr. Res. 2015, 77, 229–235. [Google Scholar] [CrossRef]
- James, K.; O’Connell Motherway, M.; Bottacini, F.; van Sinderen, D. Bifidobacterium breve UCC2003 metabolises the human milk oligosaccharides lacto-N-tetraose and lacto-N-neo-tetraose through overlapping, yet distinct pathways. Sci. Rep. 2016, 6, 38560. [Google Scholar] [CrossRef]
- Goodrich Julia, K.; Davenport Emily, R.; Beaumont, M.; Jackson Matthew, A.; Knight, R.; Ober, C.; Spector Tim, D.; Bell Jordana, T.; Clark Andrew, G.; Ley Ruth, E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef]
- Heine, W.; Mohr, C.; Wutzke, K.D. Host-microflora correlations in infant nutrition. Prog. Food Nutr. Sci. 1992, 16, 181–197. [Google Scholar]
- Pible, O.; Armengaud, J. Improving the quality of genome, protein sequence, and taxonomy databases: A prerequisite for microbiome meta-omics 2.0. Proteomics 2015, 15, 3418–3423. [Google Scholar] [CrossRef] [Green Version]
- Tanca, A.; Palomba, A.; Fraumene, C.; Pagnozzi, D.; Manghina, V.; Deligios, M.; Muth, T.; Rapp, E.; Martens, L.; Addis, M.F.; et al. The impact of sequence database choice on metaproteomic results in gut microbiota studies. Microbiome 2016, 4, 51. [Google Scholar] [CrossRef]
- Matsuki, T.; Watanabe, K.; Fujimoto, J.; Takada, T.; Tanaka, R. Use of 16S rRNA Gene-Targeted Group-Specific Primers for Real-Time PCR Analysis of Predominant Bacteria in Human Feces. Appl. Environ. Microbiol. 2004, 70, 7220–7228. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Meth. 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The Human Microbiome Project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Suzek, B.E.; Wang, Y.; Huang, H.; McGarvey, P.B.; Wu, C.H.; The UniProt Consortium. UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015, 31, 926–932. [Google Scholar] [CrossRef] [PubMed]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Annotation | #Spectral Counts 2 | # Unique Peptide Sequences 3 | # Metaclusters 4 |
|---|---|---|---|---|
| Bacterial | With useful annotation 1 | 32,469 | 10,675 | 1033 |
| Without useful annotation | 8455 | 3541 | 889 | |
| Human | With useful annotation | 8951 | 1034 | 212 |
| Subject | Spectral Counts | |||||||
|---|---|---|---|---|---|---|---|---|
| C | T | B | P | M | N | E | R | |
| Total number of strains assigned with ≥1 unique spectrum 1 | 45 | 67 | 78 | 99 | 75 | 76 | 64 | 64 |
| Number of strains assigned with ≥2 spectra | 25 | 43 | 49 | 71 | 45 | 42 | 39 | 42 |
| Number of strains assigned with ≥5 spectra | 9 | 14 | 20 | 32 | 18 | 18 | 19 | 10 |
| Subject ID | C | T | B | P | M | N | E | R |
|---|---|---|---|---|---|---|---|---|
| Gestational age (weeks) | 39 | 39 | 40 | 41 | 34 | 34 | 40 | 41 |
| Gender | M | M | M | F | M | M | F | M |
| Age (days) | 64 | 134 | 119 | 61 | 96 | 96 | 140 | 132 |
| Delivery Mode | V | V | V | CS | V | V | V | V |
| Feeding Mode | BF | BF | BF | Mixed | Mixed | Mixed | FF | BF |
| Sample weight (g) | 0.23 | 0.51 | 0.56 | 0.55 | 0.51 | 0.58 | 0.52 | 0.53 |
| Remarks | Vaccinated on sampling day (DTPP/Hib/HepB/Pneu) | Mono-zygotic twin #1 | Mono-zygotic twin #2 | Antibiotic treatment (amoxicillin) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortes, L.; Wopereis, H.; Tartiere, A.; Piquenot, J.; Gouw, J.W.; Tims, S.; Knol, J.; Chelsky, D. Metaproteomic and 16S rRNA Gene Sequencing Analysis of the Infant Fecal Microbiome. Int. J. Mol. Sci. 2019, 20, 1430. https://doi.org/10.3390/ijms20061430
Cortes L, Wopereis H, Tartiere A, Piquenot J, Gouw JW, Tims S, Knol J, Chelsky D. Metaproteomic and 16S rRNA Gene Sequencing Analysis of the Infant Fecal Microbiome. International Journal of Molecular Sciences. 2019; 20(6):1430. https://doi.org/10.3390/ijms20061430
Chicago/Turabian StyleCortes, Laetitia, Harm Wopereis, Aude Tartiere, Julie Piquenot, Joost W. Gouw, Sebastian Tims, Jan Knol, and Daniel Chelsky. 2019. "Metaproteomic and 16S rRNA Gene Sequencing Analysis of the Infant Fecal Microbiome" International Journal of Molecular Sciences 20, no. 6: 1430. https://doi.org/10.3390/ijms20061430