
Abstract
Reptiles have been shown to host a significant Helicobacter diversity. In order to survive, reptile-associated Helicobacter lineages need to be adapted to the thermally dynamic environment encountered in a poikilothermic host. The whole genomes of reptile-associated Helicobacter lineages can provide insights in Helicobacter host adaptation and coevolution. These aspects were explored by comparing the genomes of reptile-, bird-, and mammal-associated Helicobacter lineages. Based on average nucleotide identity, all reptile-associated Helicobacter lineages in this study could be considered distinct species. A whole genome-based phylogeny showed two distinct clades, one associated with chelonians and one associated with lizards. The phylogeny indicates initial adaptation to an anatomical niche, which is followed by an ancient host jump and subsequent diversification. Furthermore, the ability to grow at low temperatures, which might reflect thermal adaptation to a reptilian host, originated at least twice in Helicobacter evolution. A putative tricarballylate catabolism locus was specifically present in Campylobacter and Helicobacter isolates from reptiles. The phylogeny of reptile-associated Helicobacter parallels host association, indicating a high level of host specificity. The high diversity and deep branching within these clades supports long-term coevolution with, and extensive radiation within the respective reptilian host type.
Similar content being viewed by others
Introduction
All Helicobacter species are associated with vertebrate hosts, in which they usually colonize the mucosa of the gastrointestinal tract and the liver. Although generally believed to occur primarily in birds and mammals1, it has been shown that reptiles host a large Helicobacter diversity as well2, 3. Helicobacter occurrence in reptiles ranges from 4.8% to 39.1%, depending on the detection method used2. Based on 16S rRNA phylogeny, Helicobacter lineages isolated from reptiles formed a distinct cluster, separate from Helicobacter species isolated from mammals and birds2. This also suggested confined host association, as the lineages were separated in a cluster of isolates originating from lizards and a cluster of isolates originating from chelonians. These lineages represent up to eight putative novel species, based on 16S rRNA homology (93–98%), indicating that Helicobacter biodiversity in reptiles can be considered high, compared to the currently known species from related genera Arcobacter (three species) and Campylobacter (four species)2.
In general, whereas mammals and birds are endothermic and have more constant body temperatures (homeothermic), reptiles are ectothermic and largely dependent on external heat sources for their preferred body temperatures, which can show considerable fluctuations (poikilothermic). Consequently, Helicobacter species occurring in poikilothermic reptiles have to be adapted to larger temperature ranges and on average lower temperatures than Helicobacter species occurring in homeothermic animals.
Indeed, initial genetic and phenotypic characterization of a Helicobacter strain obtained from a western hognose snake (Heterodon nasicus) showed that this strain was distinct from other Helicobacter species in the ability to grow at lower temperatures (25 °C)4. This Helicobacter strain, for which the name Helicobacter serpensis sp. pr. (species proponenda) has been proposed, had an identical 16S rRNA sequence to Helicobacter strain 12S02232-10, which was independently isolated from a rhinoceros iguana (Cyclura cornuta), and both strains likely belong to the same species2.
No association with disease in reptiles is apparent for Helicobacter, and Helicobacter likely represents a component of the normal reptilian microbiome, although a fatal septicemia in a pancake tortoise (Malacochersus tornieri) has been shown associated with an undescribed Helicobacter species5.
The obvious host dichotomy and 16S rRNA phylogeny suggested long-term divergence and coevolution between Helicobacter and its reptilian hosts2. However, from this phylogenetic analysis it remained inconclusive whether the reptile-associated Helicobacter lineages are more related to enterohepatic or gastric Helicobacter species. Indeed, Helicobacter phylogeny based on 16S rRNA has been shown discordant with phylogenies based on other sequence data6. In this study, the genomes of Helicobacter strains from reptiles were characterized and compared to Helicobacter strains from birds and mammals to elucidate the factors contributing to adaptation to poikilothermic or homeothermic hosts, and to gain insights in Helicobacter phylogeny and the coevolutionary trajectory of Helicobacter and the reptilian host.
Results
Phylogeny of reptile-associated Helicobacter
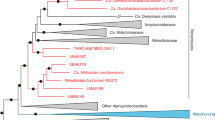
A whole genome-based phylogeny accounting for the effects of homologous recombination was reconstructed for the reptile-associated Helicobacter strains and (candidate) Helicobacter species from birds and mammals (Fig. 1). The most basal split is between the unsheathed Helicobacter species, including Wolinella succinogenes, and the other enterohepatic and gastric Helicobacter species. Notably, Wolinella succinogenes does not cluster separately from Helicobacter, but forms a separate clade together with the unsheathed Helicobacter species. The gastric Helicobacter species branch of last from the enterohepatic Helicobacter clade. The whole genome-based phylogeny suggests that the gastric Helicobacter lineages evolved most recently from an enterohepatic ancestor.

Rooted whole genome-based phylogeny for all Helicobacter strains used in this study. The squamate- and chelonian-associated Helicobacter clades are indicated with a lizard or chelonian, respectively. C. fetus strain 82–40 was used as outgroup and root.
The reptile-associated Helicobacter strains form two separate and highly divergent clades, one associated with chelonian hosts and one associated with squamate hosts (i.e. lizards and snakes). These two clades are nested within the Helicobacter genus and do not form a basal clade separate from Helicobacter species associated with avian and mammalian hosts.
The chelonian-associated Helicobacter clade is most closely related to enterohepatic Helicobacter species having periplasmic fibers, which wrap helically around the body of the bacterium and give a crisscross appearance to the bacterial surface, a morphologic feature which is often used to subdivide enterohepatic Helicobacter species1. As many enterohepatic Helicobacter species, the chelonian-associated Helicobacter strains lack the genes needed for urease production. In contrast, the squamate-associated Helicobacter clade is most closely related to gastric Helicobacter species. Similar to gastric Helicobacter species, the urease locus is conserved in all squamate-associated Helicobacter strains. A phylogeny based on urease encoding genes displayed a similar topology as the whole genome-based phylogeny (data not shown).
Speciation of reptile-associated Helicobacter
The average nucleotide identity (ANI) was calculated to determine whether the different reptile-associated Helicobacter lineages can be considered separate species. Supplementary Table S1 shows the ANI for all reptile-associated Helicobacter strains and a selection of the most closely related Helicobacter species. The ANI largely reflects the phylogeny, as the chelonian- and squamate-associated Helicobacter clades are clearly separated. All of the eight reptile-associated Helicobacter lineages show an ANI well below the species delimitation of 95–96%, which indicates that each lineage can be considered a novel species based on genetic divergence.
Thermal adaptation
The growth temperature range was determined for the reptile-associated Helicobacter strains and compared to the growth temperature range of the other bird- and mammal-associated Helicobacter species (Table 1). Notably, all reptile-associated Helicobacter strains, but none of the bird- and mammal-associated Helicobacter species, were able to grow at 25 °C. All reptile-associated Helicobacter strains showed growth at 37 °C and two of the eight reptile-associated Helicobacter strains showed growth at 42 °C.
Genetic features specific to reptile-associated Helicobacter
The genomes of all reptile-associated Helicobacter strains were screened for specific genetic features (Supplementary Table S2). A total of 17 genes, encoding mostly hypothetical proteins and two outer membrane proteins, were specific for the lizard-associated Helicobacter clade. Both chelonian-associated Helicobacter lineages specifically shared 148 genes. All reptile-associated Helicobacter lineages collectively shared three genes, encoding an acetyltransferase (GNAT) family protein, putative methyltransferase YcgJ, and glutamyl-tRNA amidotransferase subunit A. The urease locus was present in all lizard-associated Helicobacter lineages, but absent from all chelonian-associated Helicobacter lineages. Additional urease alpha and beta subunits were present in the genomes of H. acinonychis, H. cetorum, H. felis, and H. mustelae.
The amino acid sequences of each genome were searched for amino acid sequences of known H. pylori virulence factors (AlpA, AlpB, BabA, CagA, DupA, gGT, HopZ, IceA, IceA2, NapA, OipA, SabA, VacA). Most of these virulence factors (10–12) were also present in the closely related H. acinonychis and H. cetorum (Supplementary Table S3). In all other strains, including the reptile-associated Helicobacter lineages, few of these virulence factors (2–5) were present. In all reptile-associated Helicobacter lineages gGT and NapA were present, HopZ was present only in the lizard-associated Helicobacter lineages, and DupA was present in all lizard-associated Helicobacter lineages except Helicobacter 11S02596-1 and 12S02634-8.
Notably, as in reptile-associated Campylobacter lineages, genes of a putative tricarballylate catabolism locus tcuRABC were present in all reptile-associated Helicobacter lineages. Outside the reptile-associated Helicobacter lineages, these genes were only observed in the distantly related species H. canadensis and H. macacae. As the tcuRABC locus was incomplete in most lineages, the functionality may be altered or impaired.
Initial screening showed that tcuC was conserved in all reptile-associated Helicobacter and Campylobacter lineages. This gene was used to elucidate the origin of tcuC and the tcuRABC locus. A maximum likelihood dendrogram based on tcuC from all Helicobacter and Campylobacter lineages shows that tcuC from the lizard-associated Helicobacter clade is divergent from all other lineages (Fig. 2). Interestingly, tcuC from the chelonian-associated Helicobacter lineages was most closely related to tcuC from the reptile-associated Campylobacter lineages.

Rooted single gene maximum likelihood dendrogram based on tcuC from Helicobacter and Campylobacter. The chelonian- and squamate-associated strains are indicated with a chelonian or lizard, respectively. Enterobacter asburiae L1 was used as outgroup and root. Bootstrap values (≥70%) based on 500 repetitions are shown at the nodes of the dendrogram.
Discussion
Helicobacter phylogeny reflects host phylogeny only to a certain degree. In contrast to the 16S rRNA-based phylogeny, whole genome-based phylogeny shows that the reptile-associated Helicobacter lineages do not form one coherent clade. Instead, two separate and distantly related reptile-associated Helicobacter clades can be recognized; one associated with chelonians and one associated with squamates. Both clades are nested within the Helicobacter genus and do not form an apparent basal uniform sister clade to bird- and mammal-associated Helicobacter lineages. As Helicobacter phylogeny does not reflect amniotic vertebrate phylogeny, specific long-term coevolution of Helicobacter and its vertebrate host since the last common ancestor can be considered unlikely. Rather the whole genome-based phylogeny is suggestive of initial adaptation to a certain anatomical niche (e.g. gastric or intestinal), followed by a host jump and radiation in a particular host group (e.g. squamates or chelonians). This is most apparent in the squamate-associated Helicobacter clade, which is most closely related to the gastric mammal-associated Helicobacter clade, including H. pylori. The genes involved in urease production are conserved in all lineages and are indicative of a gastric niche. These genes show a similar phylogeny as the whole genome-based phylogeny, indicating long-term conservation within each lineage with an origin pre-dating the split between gastric mammal- and reptile-associated Helicobacter lineages. This is in support of an initial ancient adaptation to the gastric niche, followed by radiation in either mammals or squamates. Ancient host jumps between distantly related host-species have been reported previously for Helicobacter 7.
Interestingly, the phylogeny of reptile-associated Helicobacter parallels host association, i.e. each clade is confined to a phylogenetically distinct host type (either chelonian or squamate), indicating a high level of host specificity. In combination with the observed high diversity and deep branching within these clades, this supports long-term coevolution with, and extensive radiation within the respective reptilian host type.
Within the squamate-associated Helicobacter clade, Helicobacter strains obtained from recently wild-caught lizards from the same original geographic region clustered together, indicating an association with geographic origin. This association was not apparent amongst the other squamate-associated Helicobacter strains. As these were obtained from captive-held animals this signal could be obscured due to anthropogenic influences. Interspecies transmission in unnatural animal assemblies have been noted before for Campylobacter and Helicobacter 2, 3. Occurrence in multiple host types has been observed in reptile-associated Helicobacter as well, as an identical Helicobacter lineage was isolated from a captive-held lizard and a snake. However, the presence of Helicobacter in recently wild-caught reptiles, combined with the phylogenetic coherence, suggests that Helicobacter is likely a relevant constituent of the gastrointestinal microbiome in wild reptiles as well.
Growth at low temperature (≤25 °C) is observed in all reptile-associated Helicobacter lineages, but is not known from any Helicobacter species isolated from either birds or mammals. This shows that growth at low temperature is entirely associated with occurrence in a reptilian host. As this feature is observed in both distantly related chelonian- and squamate-associated Helicobacter clades, adaptation to the variable but on average low temperatures encountered in a poikilothermic reptilian host originated independently and at least twice in Helicobacter evolution. The observation that some squamate-associated Helicobacter lineages were able to grow at 42 °C and show the largest growth temperature range known for Helicobacter confirms the large thermal adaptation needed to survive in a poikilothermic host.
Genes putatively involved in tricarballylate catabolism (tcuRABC) were present in all reptile-associated Helicobacter lineages. Outside the reptile-associated Helicobacter lineages tcuRABC genes are only found in H. canadensis and H. macacae. In the closely related genus Campylobacter these genes are also predominantly found in the reptile-associated taxa C. fetus subsp. testudinum and C. iguaniorum 8, 9. This indicates that these genes may be important in survival in a reptilian host. As has been shown for Salmonella enterica, these genes potentially enable reptile-associated Helicobacter and Campylobacter lineages to use the citrate analog tricarballylate as carbon and energy source, which may provide an advantage for survival in a reptilian host10. Tricarballylate is toxic to ruminants by inhibiting aconitase and the citric acid cycle11. However, reptiles have been shown less susceptible to aconitase inhibition than mammals12. As such, reptiles are expected to be more tolerant to tricarballylate, which might be more abundant in the reptilian than in the mammalian gastrointestinal tract. Noteworthy, tcuC from chelonian-associated Helicobacter lineages is closer related to tcuC from reptile-associated Campylobacter lineages than to tcuC from lizard-associated Helicobacter lineages. This suggests lateral transfer of tcuC between Helicobacter and Campylobacter, potentially in a chelonian host, which shows the highest Campylobacter prevalence amongst reptiles2.
Based on our results, and in contrast to 16S rRNA-based phylogeny (Supplementary Figure S1), Wolinella succinogenes forms a clade together with Helicobacter species having unsheathed flagella. Excluding W. succinogenes from the Helicobacter phylogeny would leave it paraphyletic, which implies that W. succinogenes could be considered a member of the Helicobacter genus. Based on 16S rRNA, all reptile-associated Helicobacter lineages, both urease positive and negative, form a distinct clade together with H. mustelae and urease negative H. pametensis. As shown previously for Helicobacter, phylogenies based on 16S rRNA are discordant with 23S rRNA-based phylogenies and other data, which is consistent with the horizontal transfer of 16S rRNA gene fragments and loss of phylogenetic information6. As such, 16S rRNA might be less suitable for phylogenetic analysis of Helicobacter.
Based on the ANI values, all reptile-associated Helicobacter lineages included in this study represent novel species. With eight putative species, the diversity of Helicobacter in reptiles is high compared to the other vertebrate-associated Epsilonproteobacteria genera Arcobacter and Campylobacter (three and four species, respectively)2. An explanation of the high Helicobacter diversity could be a higher host or niche specificity, leading to more isolation, thereby facilitating diversification. Also, a more ancient introduction of Helicobacter in a reptilian host could have led to more extended diversification.
It has to be noted that several members of the Helicobacter genus are considered fastidious micro-organisms and likely many more Helicobacter lineages are present in reptiles than the ones included in this study, which may also include enterohepatic Helicobacter in squamates and gastric Helicobacter in chelonians. Furthermore, as all isolates were obtained from intestinal contents from cloacal swabs, the exact region of the gastrointestinal tract colonized by the reptile-associated Helicobacter isolates included in this study is not known, but rather the presumed anatomical niche is inferred from the position in the phylogenetic tree and the presence or absence of the urease locus. More culturing- and sequencing-based studies are needed to provide further insights in the exact diversity, phylogeny, and niche preference of Helicobacter in reptiles.
In conclusion, poikilothermic reptiles host a large diversity of Helicobacter lineages, which are distinct from bird- and mammal-associated Helicobacter species. These reptile-associated Helicobacter lineages provide novel insights in Helicobacter host adaptation, phylogeny, and evolution. Given the large diversity of Helicobacter in a limited number of well-investigated host species, it is expected that the total Helicobacter diversity in vertebrates far exceeds the currently known diversity. In all probability, further sampling of other reservoirs, preferably wild animals, should lead to an increase of Helicobacter diversity and a further refinement of Helicobacter phylogeny.
Methods
Strains
Helicobacter strains representing eight putative novel species were isolated from intestinal contents from cloacal swabs of chelonians and lizards as described previously2. All strains were isolated from captive-held animals from zoos, pet shops or private collections. None of the hosts had apparent intestinal illness or other clinical signs. By default, strains were grown on Columbia agar with 5% sheep blood (Oxoid, the Netherlands) in a microaerobic atmosphere (83.3 N2, 7.1% CO2, 3.6% H2, and 6% O2) at 37 °C for 48 h. To determine growth temperature range, the strains were also grown at 25 °C and 42 °C. Characteristics of all strains used in this study are summarized in Table 1.
Whole genome sequencing
Sequencing of the reptile-associated Helicobacter strains was performed using Illumina MiSeq, 300 bp read length. The reads were assembled using SPAdes 3.1.1. The average coverage was 212× and average number of contigs was 59. The whole genome sequences of all reptile-associated Helicobacter strains have been deposited at GenBank. All available whole genome sequences of other (candidate) Helicobacter and Wolinella species were extracted from GenBank on March 9th 2016. Genomic features and accession numbers of all Helicobacter and Wolinella genomes used in this study can be found in Supplementary Table S3.
In addition to this, the whole genome sequences of Enterobacter asburiae L1 (GenBank accession number CP007546.1), Campylobacter coli N11D1 (FBQY00000000.1), C. cuniculorum DSM 23162 (JHZL00000000.1), C. fetus subsp. fetus 82–40 (CP000487.1), C. fetus subsp. testudinum 03–427 (CP006833.1), C. iguaniorum 1485E (CP010995.1), and C. jejuni OXC6631 (CUVR00000000.1) were used in this study.
Genome analysis
For prokaryote species delineation, the average nucleotide identity (ANI) can be used as an alternative for DNA-DNA hybridization (DDH)13, 14. A DDH species delineation of 70% corresponds to about 95% ANI15. Using the JSpecies ANI tool16, pair-wise ANI values based on the whole genome sequences were calculated for all strains used in this study.
To determine the presence of virulence factors, amino acid sequences of known H. pylori virulence factors (AlpA, AlpB, BabA, CagA, DupA, gGT, HopZ, IceA, IceA2, NapA, OipA, SabA, VacA) were aligned against the predicted amino acid sequences of each genome at an e-value cutoff of 1E-50 (1E-30 for IceA2) using BLAST.
Orthologous grouping and phylogenomic reconstruction
An all versus all BLAST was performed for all predicted proteins of the whole genomes (Table 1) at an E-value cutoff of 1E-6. To determine the orthologous relationships of all proteins, the BLAST output was parsed by Orthagogue using default settings17. To determine the orthologous groups, Markov clustering (MCL) was performed using MCL-edge18. Genes encoding the proteins were aligned with each other within their respective orthologous groups using MUSCLE19. A super alignment of 603,413 nt was created by concatenating the aligned genes according to their position in H. pylori J99 if they were present in all isolates. Gaps were removed using TrimAl resulting in a 302,243 nt super alignment20. Based on this super alignment phylogenomic reconstruction and prediction of recombination events was performed using Gubbins21 with the default settings. Whole genome phylogeny was based on a gapless super alignment. Phylogenetic dendrograms were created using Fasttree22 or MEGA 623.
For the 16S rRNA-based phylogeny, the 16S rDNA sequences were extracted from the genomes, aligned using MUSCLE19, gap positions and flanking sequences were removed resulting in a 1290 bp alignment. A phylogenetic tree was constructed using RaxML24 with a GTR model with gamma correction. The tree was rooted on C. fetus 82–40 and visualized using Figtree.
Data availability
The data that support the findings of this study are included in this published article (and its Supplementary Information files) and are available in the GenBank database.
References
Schauer, D. B. In Helicobacter pylori: Physiology and genetics (eds Mobley, H. L. T., Mendz, G. L. & Hazell, S. L.) (ASM Press, Washington (DC), 2001).
Gilbert, M. J. et al. Occurrence, diversity, and host association of intestinal Campylobacter, Arcobacter, and Helicobacter in reptiles. PLoS One 9, e101599 (2014).
Schrenzel, M. D. et al. Genetic characterization and epidemiology of Helicobacters in non‐domestic animals. Helicobacter 15, 126–142 (2010).
Lawson, A. & Owen, R. Helicobacter serpensis, a novel Helicobacter species isolated from snake faeces (Zoonoses and public health Ser. 54, Blackwell Publishing, England, 2007).
Stacy, B. A. & Wellehan, J. F. Jr. Fatal septicemia caused by Helicobacter infection in a pancake tortoise (Malacochersus tornieri). J. Vet. Diagn. Invest. 22, 660–662 (2010).
Dewhirst, F. E. et al. Discordant 16S and 23S rRNA gene phylogenies for the genus Helicobacter: implications for phylogenetic inference and systematics. J. Bacteriol. 187, 6106–6118 (2005).
Eppinger, M. et al. Who ate whom? Adaptive Helicobacter genomic changes that accompanied a host jump from early humans to large felines. PLoS Genet 2, e120 (2006).
Gilbert, M. J. et al. Comparative genomics of Campylobacter fetus from reptiles and mammals reveals divergent evolution in host-associated lineages. Genome Biol Evol 8, 2006–2019 (2016).
Gilbert, M. J. et al. Comparative genomics of Campylobacter iguaniorum to unravel genetic regions associated with reptilian hosts. Genome Biol Evol 8, 3022–3029 (2016).
Lewis, J. A., Horswill, A. R., Schwem, B. E. & Escalante-Semerena, J. C. The tricarballylate utilization (tcuRABC) genes of Salmonella enterica serovar Typhimurium LT2. J. Bacteriol. 186, 1629–1637 (2004).
Russell, J. B. Enrichment and isolation of rumen bacteria that reduce trans-aconitic acid to tricarballylic acid. Appl. Environ. Microbiol. 49, 120–126 (1985).
McIlroy, J. The sensitivity of Australian animals to 1080 poison. 9. Comparisons between the major groups of animals, and the potential danger nontarget species face from 1080 poisoning campaigns. Wildl. Res. 13, 39–48 (1986).
Konstantinidis, K. T. & Tiedje, J. M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 102, 2567–2572 (2005).
Konstantinidis, K. T., Ramette, A. & Tiedje, J. M. The bacterial species definition in the genomic era. Philosophical Transactions of the Royal Society B: Biological Sciences 361, 1929–1940 (2006).
Goris, J. et al. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91 (2007).
Richter, M. & Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 106, 19126–19131 (2009).
Ekseth, O. K., Kuiper, M. & Mironov, V. OrthAgogue: an agile tool for the rapid prediction of orthology relations. Bioinformatics 30, 734–736 (2014).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30, 1575–1584 (2002).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Croucher, N. J. et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43 (2014).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Haesebrouck, F. et al. Gastric helicobacters in domestic animals and nonhuman primates and their significance for human health. Clin. Microbiol. Rev. 22, 202–223 (2009).
Acknowledgements
We thank Andy Lawson for initial characterization of Helicobacter strain SP2.
Author information
Authors and Affiliations
Contributions
A.Z., M.G., and B.D. performed the analyses. A.T. performed technical support. M.G. wrote the manuscript. M.G., B.D., A.Z., and J.W. designed the study and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gilbert, M.J., Duim, B., Timmerman, A.J. et al. Whole genome-based phylogeny of reptile-associated Helicobacter indicates independent niche adaptation followed by diversification in a poikilothermic host. Sci Rep 7, 8387 (2017). https://doi.org/10.1038/s41598-017-09091-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09091-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
