
Abstract
Proteins are vital in all biological systems as they constitute the main structural and functional components of cells. Recent advances in mass spectrometry have brought the promise of complete proteomics by helping draft the human proteome. Yet, this commonly used protein sequencing technique has fundamental limitations in sensitivity. Here we propose a method for single-molecule (SM) protein sequencing. A major challenge lies in the fact that proteins are composed of 20 different amino acids, which demands 20 molecular reporters. We computationally demonstrate that it suffices to measure only two types of amino acids to identify proteins and suggest an experimental scheme using SM fluorescence. When achieved, this highly sensitive approach will result in a paradigm shift in proteomics, with major impact in the biological and medical sciences.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Corrections were made to this article on 7 February 2017. Corrected supplementary data has been uploaded.
In 2014 two international teams produced the first draft of the human proteome, using mass spectrometry (MS) [1, 2]. By opening a new chapter in proteomics, these large scale studies will help us understand complex cellular processes. Yet, MS—the most widely used protein sequencing technology—requires a large amount of sample. This hampers quantification, precludes detecting many proteins of interest that are present only in low concentrations in the cell, and renders single-cell analysis impossible.
Single-molecule (SM) protein sequencing would bring about 'protein deep sequencing' [3–5]. However, unlike DNA sequencing that needs to read out only four nucleotides, protein sequencing demands differentiation of 20 amino acids, far beyond what current SM techniques can offer [3]. SM protein sequencing has therefore not followed up SM DNA sequencing that uses fluorescence and nanopores [6–8]. Here we propose a novel SM protein sequencing method that overcomes this challenge and assess its feasibility using computational analysis.
Unique to protein sequencing is that a protein can be identified using incomplete information with reference to proteomic databases. Consider a 2 bit fingerprinting scheme in which only two types of amino acids are labeled (figure 1). A consecutive read of 15 labeled amino acids is sufficient to identify up to 215 = 32 768 unique protein sequences. This exceeds the number of (major isoform) protein species that most organisms express. As the median length of a protein ranges from 270 (bacteria) to 350 amino acids (eukaryotes), it is not difficult to choose two amino acid types that appear more than 15 times in each protein (supplementary figure 1, stacks.iop.org/PB/12/055003/mmedia).
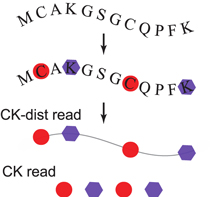
Figure 1. A single-molecule read-out. CK fingerprinting read: the order of C's and K's are detected. CK-dist fingerprinting read: the distances between C's and K's are additionally measured.
Download figure:
Standard image High-resolution imageFigure 2 describes a SM protein fingerprinting scheme using fluorescence. We chose to label two highly nucleophilic amino acids, lysine (K) and cysteine (C) as they are frequent (supplementary figure 2) and can be labeled both efficiently and orthogonally (NHS–ester coupling with lysine and maleimide coupling with cysteine) [9]. A similar idea using lysine and arginine for monitoring protein synthesis inside a living cell was patented by Anima Cell Metrology [10]. Recently, Swaminathan et al discussed fingerprinting schemes that are based on multiple labels, including two labels [3]. Separately, a work published in 2013 shows how our fingerprinting approach might be implemented using nanopores [4].
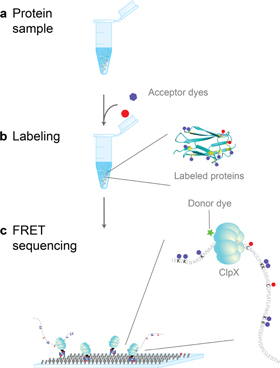
Figure 2. The schematic shows how single-molecule fingerprinting is carried out. (a) Proteins are obtained from diverse sources including single cells, tissues, and body fluids. (b) Extracted proteins are denatured, and cysteines and lysines are labeled with fluorescent dyes. (c) An engineered version of a protein translocase (e.g. bacterial ClpX) grabs individual substrate proteins, unfolds them, and translocates them through its nanochannel. Proteins are sequenced using FRET (Förster resonance energy transfer). The translocase is labeled with a donor dye. FRET occurs between the donor on the translocase and the two distinct acceptor dyes on a substrate when the substrate passes through the nanomachine. The FRET signals report the order of the labeled amino acids.
Download figure:
Standard image High-resolution imageTo assess the predictive power of fingerprinting, we developed a dedicated search algorithm. In brief, we search CK (cysteine–lysine) fingerprints by combining a filtering strategy to decrease computation time with a dynamic programming-based alignment step, considering a specific set of potential experimental errors (see methods).
For our analysis, we used a canonical human proteome database based on Uniprot release 2014.04 [11]. We simulated 2000 different read-outs, searched for each of them in the database and measured the detection precision (P), i.e. the probability of retrieving the correct sequence. In an ideal situation with no experimental error, P is 90% (figure 3(a), blue). Next, we assessed the robustness of the method against inaccuracies that are expected from actual experiments, by iteratively introducing errors into each fingerprint at random (see error simulation in methods for details) up to a specified error level ( a number of errors (
a number of errors ( divided by a CK fingerprint length (lck, the length of CK sequences excluding other amino acids). Figure 3 reports P of these computations. As expected, P drops when
divided by a CK fingerprint length (lck, the length of CK sequences excluding other amino acids). Figure 3 reports P of these computations. As expected, P drops when  increases. For example, at
increases. For example, at  half of the sequences are correctly and uniquely retrieved (figure 3(a), blue).
half of the sequences are correctly and uniquely retrieved (figure 3(a), blue).
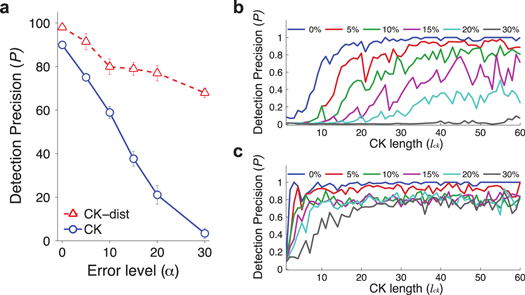
Figure 3. (a) Detection precision, P, at various error levels,  blue for CK fingerprinting, red for CK-dist fingerprinting. Error bars are the standard deviation from three independent simulations. (b) P as a function of CK fingerprint length (lck, the length of CK sequences excluding other amino acids) at various
blue for CK fingerprinting, red for CK-dist fingerprinting. Error bars are the standard deviation from three independent simulations. (b) P as a function of CK fingerprint length (lck, the length of CK sequences excluding other amino acids) at various  for CK fingerprinting. (c) P as a function of lck at various
for CK fingerprinting. (c) P as a function of lck at various  for CK-dist fingerprinting.
for CK-dist fingerprinting.
Download figure:
Standard image High-resolution imageTo improve performance, we considered other information: the distance between C's and K's (figure 3(a), red). At any  P was dramatically higher with the distance included: at
P was dramatically higher with the distance included: at  P increased to 85%. In general, P increases when lck becomes longer (figures 3(b)–(c)). At any lck, P for CK fingerprinting with distance information (named 'CK-dist fingerprinting') is higher than or equal to P for CK fingerprinting. A similar observation was made when different additional information was considered (supplementary figure 5). Taken together, these demonstrate the feasibility of the technique for identifying primary protein sequences.
P increased to 85%. In general, P increases when lck becomes longer (figures 3(b)–(c)). At any lck, P for CK fingerprinting with distance information (named 'CK-dist fingerprinting') is higher than or equal to P for CK fingerprinting. A similar observation was made when different additional information was considered (supplementary figure 5). Taken together, these demonstrate the feasibility of the technique for identifying primary protein sequences.
Another application area could be clinical diagnostics. As an example of detecting infections, we chose human respiratory syncytial virus (HRSV) and tuberculosis (TB). We determined that a set of HRSV and TB proteins contain a unique CK fingerprint and thus can be detected at α as high as 15–20% and be potentially used as markers for HRSV and TB (supplementary figure 6).
The CK fingerprinting technique will also enable us to detect post-translational modifications of proteins when it is expanded to a three-color fluorescence measurement. For example, glycosylated amino acids can be labeled with a third acceptor dye using hydrazide–aldehyde coupling chemistry, which is orthogonal to the labeling methods for lysine and cysteine residues. Phosphorylated serine and threonine can be labeled with a third acceptor using another coupling scheme [12]. This will advance the proof-of-principle of detecting a post-translationally modified peptide using a nanopore that was reported in 2014 [5].
We described SM protein fingerprinting, a technique that will provide proteomics with high sensitivity and a large dynamic range. Our computational assessment indicated that, even if we read only two amino acid types, we could correctly identify proteins with reference to proteomic databases. When this entirely new SM protein sequencing approach is achieved, it will become a proteomics tool that complements MS and opens up new avenues in global, high-throughput protein analysis.
Methods
Here we describe the approaches that we used to simulate errors and find protein fingerprints that match a given query fingerprint pattern.
Error simulation
We simulated 2000 read-outs, each for a different protein. The proteins are randomly picked from the database and thus contain random amino acids and fingerprint lengths. Next, to assess the robustness of the method against inaccuracies that are expected from actual experiments, errors are iteratively introduced for each read-out up to the error level we want to investigate.
We expect that actual data will be convoluted with poor dye-labeling, photoblinking and photobleaching of dyes, local structures of a substrate protein, non-uniform speed of substrate translocation, proximity between dyes etc. The poor labeling, photoblinking, and photobleaching of acceptor dyes will appear as deletion errors (figure 4). The non-uniform speed of translocation will introduce insertion and deletion errors to CK-dist fingerprinting. The proximity of acceptor dyes will bring deletion and transposition/substitution errors. If a donor dye is photobleached during a measurement, it will appear as a truncation error. We do not consider this error for fingerprinting analysis since donor photobleaching can be determined from SM time traces and thus can be easily excluded from further analysis. Other complications, such as aggregation of denatured proteins, may also be expected but are not considered in our analysis. See a pseudo-code for simulating these errors (supplementary information).

Figure 4. Expected experimental errors. 'R' is reference sequence. 'Q' is query sequence.
Download figure:
Standard image High-resolution imageIn figure 3, we investigated one combination of errors (70% deletions, 20% insertions, 10% transpositions) for CK fingerprinting, in which we assigned the largest percentage to deletions since this error is the most likely to occur (poor labeling of acceptors due to incomplete denaturation of proteins, photoblinking/photobleaching of acceptors, and presence of consecutive identical acceptor fluorophores). For CK-dist fingerprinting analysis, we considered the same combination of errors as for CK fingerprinting but with errors at CK residues and errors of the distance between CK residues equally likely to occur. In supplementary figure 3, we expanded the error space that we explored and obtained trends nearly identical to that found in figure 3(a).
Overview of the CK fingerprinting
The 2000 simulated readouts are searched for in the database, and the numbers of true positives and the number of matches are recorded. To examine the performance variability of our algorithm in retrieving proteins using fingerprints, three independent repetitions are executed. In each repetition, detection precision (P) (figure 3) and detection recall (R) (supplementary figure 4) are calculated based on the outputs. P is defined as the number of true positives divided by the number of read-outs returned by the algorithm. R is the number of true positives divided by the number of conditional matches.
The inputs to our method are a reference database  containing fingerprint representations of protein sequences, a query fingerprint
containing fingerprint representations of protein sequences, a query fingerprint  and an error level
and an error level  The alphabet is
The alphabet is  since we only compare fingerprints of these two amino acids. Let
since we only compare fingerprints of these two amino acids. Let  be the query length and
be the query length and  denote the
denote the  th reference sequence in the database
th reference sequence in the database  with length
with length  The distance
The distance  between a reference fingerprint
between a reference fingerprint  and a query
and a query  is the minimal number of steps required to transform
is the minimal number of steps required to transform  into
into  . Formally, given
. Formally, given 
 and
and  the problem is to find all
the problem is to find all  for which
for which  is smaller than
is smaller than  =
=  .
.
Given the inputs, the algorithm takes two steps to retrieve matches: (1) a filtration strategy is applied to identify candidate sequences in  and (2) a verification method is employed to examine all candidates for possible matches.
and (2) a verification method is employed to examine all candidates for possible matches.
Filtration: eliminating uninteresting sequences
Dynamic programming is computationally costly, prohibiting direct application on large databases in a high-throughput setting [13]. In order to reduce the running time without affecting sensitivity, we use filtration to remove those references that definitely cannot match the query fingerprint  with distance smaller than or equal to
with distance smaller than or equal to  Filtration exploits the fact that it is easier to tell a reference fingerprint that does not match a query fingerprint than to tell one that does match. Typically, it uses a simple and highly efficient filter criterion to analyze the reference sequences, leaving only a small number of
Filtration exploits the fact that it is easier to tell a reference fingerprint that does not match a query fingerprint than to tell one that does match. Typically, it uses a simple and highly efficient filter criterion to analyze the reference sequences, leaving only a small number of  for further (more expensive) analysis. We devised a new filtration method combining two existing algorithms, partial exact matching and
for further (more expensive) analysis. We devised a new filtration method combining two existing algorithms, partial exact matching and  counting.
counting.
In partial exact matching, the query fingerprint  is divided into
is divided into  pieces
pieces  where
where  equals
equals  For a match to be possible, there must be at least one piece that appears exactly in a reference sequence
For a match to be possible, there must be at least one piece that appears exactly in a reference sequence  [15]. If this is not the case,
[15]. If this is not the case,  is discarded.
is discarded.
A faster filtration method is  counting, which compares the
counting, which compares the  of two fingerprints. An
of two fingerprints. An  [16] on the alphabet set
[16] on the alphabet set  is any string in
is any string in  where
where  is the set of all possible strings of length
is the set of all possible strings of length  over
over  For example, the
For example, the  for
for  are CC, CK, KC and KK. The
are CC, CK, KC and KK. The  distance is defined as the sum of the absolute differences between the numbers of occurrences of each
distance is defined as the sum of the absolute differences between the numbers of occurrences of each  If the
If the  distance exceeds
distance exceeds 
 is discarded [16].
is discarded [16].
We combined the partial exact matching and  counting approaches to decide whether there exists at least one piece in
counting approaches to decide whether there exists at least one piece in  that appears with a limited amount of errors as a piece of
that appears with a limited amount of errors as a piece of  [17]. The distance function between two pieces of
[17]. The distance function between two pieces of  and
and 
 and
and  based on their
based on their  was defined as:
was defined as:
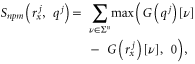
where ![$\left[\nu \right]$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn66.gif) is an
is an  and
and ![$G\left({q}^{j}\right)\left[\nu \right]$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn68.gif)
![$\text{and}\;G\left({r}_{x}^{j}\right)\left[\nu \right]$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn69.gif) denote the total number of times
denote the total number of times ![$\left[\nu \right]$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn70.gif) occurs in
occurs in  and
and  respectively.
respectively.
For each piece  in the query, the corresponding piece
in the query, the corresponding piece  contains the same letters in the reference sequence with an additional
contains the same letters in the reference sequence with an additional  letters on both sides, as shown in figure 5. It is sufficient to compare the
letters on both sides, as shown in figure 5. It is sufficient to compare the  in the reference with the
in the reference with the  in the query to determine whether the piece
in the query to determine whether the piece  appears in the reference
appears in the reference  since
since  errors cannot alter more than
errors cannot alter more than  positions. Since a query piece is searched in a limited range in the reference, it can discard more entries in the reference database than the partial exact matching method, in which the
positions. Since a query piece is searched in a limited range in the reference, it can discard more entries in the reference database than the partial exact matching method, in which the  is compared with the entire reference sequence.
is compared with the entire reference sequence.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
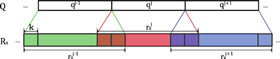
Figure 5. Consecutive pieces 
 and
and  of query
of query  and their corresponding pieces
and their corresponding pieces 
 and
and  in reference
in reference  Each query piece is compared to a limited range in the reference.
Each query piece is compared to a limited range in the reference.
Download figure:
Standard image High-resolution image{kind=link}
The distance between a piece  in query
in query  and the corresponding piece
and the corresponding piece  in
in  is computed to determine whether
is computed to determine whether  is a candidate match. For each
is a candidate match. For each  and its corresponding
and its corresponding  we check whether any
we check whether any  occurs more often in
occurs more often in  than in
than in  If not, the
If not, the  is zero, i.e. the n-grams in
is zero, i.e. the n-grams in  appear exactly in
appear exactly in  . Only if for at least one
. Only if for at least one 
 is zero,
is zero,  is kept as a candidate.
is kept as a candidate.
Verification: finding matches
The remaining candidate matches are examined by a global alignment dynamic programming approach considering a number of possible error types. In our analysis, four types of error may occur: deletion, insertion, mismatching an amino acid with another one (substitution), and swapping (transposition).
The dynamic programming algorithm is designed to provide the optimal gapped alignment between two sequences, i.e. an alignment with long regions of identical amino acid pairs and very few mismatches and gaps [14]. As the sequences become more dissimilar, more mismatched amino acid pairs and gaps should appear. To find the optimal alignment, a dynamic programming matrix  first needs to be calculated. Each element
first needs to be calculated. Each element  represents the maximum score of aligning the substrings
represents the maximum score of aligning the substrings ![$Q[\mathrm{1..}.i]$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn109.gif) and
and ![${R}_{x}[\mathrm{1..}.j].$](https://content.cld.iop.org/journals/1478-3975/12/5/055003/revision1/pb517144ieqn110.gif) Let
Let  denote the scores of the four operations. The base cases,
denote the scores of the four operations. The base cases,  and
and  are defined as (
are defined as ( and (
and ( for all
for all  (length of
(length of  and
and  (length of
(length of  respectively. Then, considering the four possibilities,
respectively. Then, considering the four possibilities,  is updated using the following recursive relation
is updated using the following recursive relation
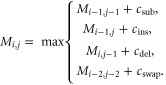
The score for each operation is set based on the estimation of how likely each error is to occur in our measurements. Currently, deletions caused by low labeling efficiency are the dominating errors, followed by insertions, transpositions and substitutions (i.e. matching C to K or vice versa) (see error simulation). Hence we choose a relatively low penalty (negative) for deletions and higher penalties (negative) for transpositions and substitutions. For the matching positions, the score is positive (see supplementary table 2).
By memorizing the solutions to the subproblems for  and
and  stored in the dynamic matrix, we can recursively compute the maximum score of aligning
stored in the dynamic matrix, we can recursively compute the maximum score of aligning  and
and  Therefore we find the score of the optimal alignment of the two sequences starting from the maximum value in the last row or last column. We maintain a matrix of traceback pointers in the recursion, so that we remember which case was used to calculate every cell
Therefore we find the score of the optimal alignment of the two sequences starting from the maximum value in the last row or last column. We maintain a matrix of traceback pointers in the recursion, so that we remember which case was used to calculate every cell  allowing to reconstruct the optimal alignment.
allowing to reconstruct the optimal alignment.
From this alignment the numbers of errors for different types as well as the total number of errors can be calculated. The distance between the query and the reference  is defined as the total number of errors. If this distance is smaller than
is defined as the total number of errors. If this distance is smaller than  the reference sequence
the reference sequence  is considered as a match. Otherwise, it is not a match of the query sequence within the error bound
is considered as a match. Otherwise, it is not a match of the query sequence within the error bound  A match is considered a true positive match when the match is the exact query protein. If a match has the same fingerprint but a different amino acid sequence, it is not considered to be a true positive match. In our analysis, this is determined by checking the protein accession codes.
A match is considered a true positive match when the match is the exact query protein. If a match has the same fingerprint but a different amino acid sequence, it is not considered to be a true positive match. In our analysis, this is determined by checking the protein accession codes.
Additional information, such as the distance between two read-outs, can be deduced from the measurements. This distance is the space between two labeled amino acids, which is the number of non-labeled amino acids in between, which show a different pattern in the measurement. For this to be estimated reliably, proteins will have to be sequenced at a relatively constant speed, an assumption which is not a priori valid. From the sequencing signals, we cannot easily determine the start or the end of proteins in the time trace if they do not correspond to a labeled amino acid. Thus, the starting and ending non-labeled amino acids are not included when we construct the fingerprint with distance information.
This distance information is added to the original CK fingerprints using an additional symbol (say, 'o'), occurring multiple times (representing the length of distance). Next, two distances between query and reference are calculated to examine whether a reference sequence is a match. One is the  between fingerprints with distance information, the other the
between fingerprints with distance information, the other the  between CK fingerprints only. Reference sequence
between CK fingerprints only. Reference sequence  is considered a match if and only if
is considered a match if and only if  is smaller than
is smaller than  and
and  is smaller than
is smaller than  where
where  is the length of the query CK fingerprint,
is the length of the query CK fingerprint,  is the length of the query fingerprint with distance information and
is the length of the query fingerprint with distance information and  and
and  represent the numbers of errors allowed. Experimental error on the distance information is also taken into consideration.
represent the numbers of errors allowed. Experimental error on the distance information is also taken into consideration.
Note: Supplementary information is available in the online version of the paper. An animated experimental scheme is available at https://youtube.com/watch?v=YpWCCWO5q10.
Acknowledgments
We would like to thank L Restrepo and L Loeff for critical reading. C J was funded by Foundation for Fundamental Research on Matter (12PR3029).
Author contributions
C J conceived the study. Y Y, M D, and D R conducted the computational analysis. Y Y, M D, J G, D R and C J discussed the data and wrote the manuscript.
Competing financial interests
C J and J G filed a patent (WO2014014347).