
Abstract
An approach based on Gateway recombination technology to efficiently construct silencing vectors was developed for use in the biotechnologically important fungus Aspergillus niger. The transcription activator of xylanolytic and cellulolytic genes XlnR of A. niger was chosen as target for gene silencing. Silencing was based on the expression vector pXLNRir that was constructed and used in co-transformation. From all the strains isolated (N = 77), nine showed poor xylan-degrading activities in two semi-quantitative plate assays testing different activities for xylan degradation. Upon induction on d-xylose, transcript levels of xlnR were decreased in the xlnR-silenced strains, compared to a wild-type background. Under these conditions, the transcript levels of xyrA and xynB (two genes regulated by XlnR) were also decreased for these xlnR-silenced strains. These results indicate that the newly developed system for rapid generation of silencing vectors is an effective tool for A. niger, and this can be used to generate strains with a tailored spectrum of enzyme activities or product formation by silencing specific genes encoding, e.g., regulators such as XlnR.
Similar content being viewed by others


Introduction
Aspergillus niger is a filamentous fungus with prominent applications in biotechnological processes. Products of the fungus have the generally recognized as safe (GRAS) status allowing them to be used as food additives. Important products that are made by fermentation of A. niger are organic acids, such as citric acid and gluconic acid. Citric acid is used for acidification and to enhance taste and flavour in food (Ruijter et al. 2000). A. niger is also an important producer of enzymes that are used in food and feed applications, such as xylanases as a bread improver and pectinases in juice clarification.
Genetic and genomic tools have been developed for this fungus over the past two decades, allowing specific modification and strain improvement to enhance the production of specific enzymes. The potential use of new genomic tools has increased with the publication of the genome sequence of A. niger (Pel et al. 2007). Recently, successful gene knockout strategies were carried out in strains with defective pathways for non-homologous integration (Meyer et al. 2007). However, these rely on a mutant background, and analysis of essential genes is still not straightforward, since it is limited to heterokaryon formation in the case of A. niger. Moreover, as reviewed (Meyer 2008), these engineered strains are more susceptible to toxins and irradiation, probably because Ku proteins have a vital role in maintenance of telomere length and chromosome stability. In addition, because this method needs a mutant background, it is not applicable toward industrial strains.
In recent years, gene silencing mediated by double-stranded (ds)RNA has opened new possibilities for the study of gene function in many organisms. Starting with pioneer studies on post-transcriptional gene silencing (PTGS) phenomena (Napoli et al. 1990; van der Krol et al. 1990; Romano and Macino 1992), many aspects of PTGS by dsRNA have been elucidated after the first study reporting the concept of dsRNA-mediated gene silencing (Fire et al. 1998).
In fungi, a number of genes involved in silencing were first described for Neurospora crassa (Cogoni and Macino 1997) and included QDE-1 (Cogoni and Macino 1999a), QDE-2 (Catalanotto et al. 2000), QDE-3 (Cogoni and Macino 1999b) and DCL1 (sms-3)/DCL2 (Catalanotto et al. 2004). Since then, many vectors for RNA-mediated gene silencing in fungi have been used successfully (Goldoni et al. 2004; Fitzgerald et al. 2004; Mouyna et al. 2004; Nakayashiki et al. 2005). Interestingly, there is indication of occurrence of small interfering RNA-mediated mechanisms in A. niger (Barnes et al. 2008).
In some cases, the use of RNA interference (RNAi) technologies may be advantageous compared to other gene-targeting techniques. “”First, for the study of essential genes since RNAi results in down-regulation of the gene function and not in loss-off-function, as is the case with a knockout strategy. Second, multiple copies of a gene or paralogous genes may be targeted with a single construct. Third, if the transformation system is based on a dominant selection marker as, e.g., the amdS gene, a method inducing gene down-regulation can be used in industrial strains.
Functional genomics based on large-scale RNAi screens has been attempted for many organisms (Berns et al. 2004; Paddison et al. 2004; Kamath et al. 2003; Boutros et al. 2004). The application of RNAi high-throughput screens to the study of gene functions in filamentous fungi, however, is still in its initial phase.
In A. niger, the expression of the biotechnologically important xylanase and cellulase enzymes is controlled by the Gal4-type transcription factor XlnR (van Peij et al. 1998b). XlnR controls not only the transcription of more than ten genes involved in the extracellular degradation of cellulose and hemi-cellulose (van Peij et al. 1998a) but also the transcription of genes that are involved in intracellular d-xylose metabolism (Hasper et al. 2000). The XlnR regulon has extensively been studied by Northern blot analysis (van Peij et al. 1998a), and many aspects of XlnR-regulated expression of target genes are known. Standardised culture and induction conditions have been developed that could serve as a basis to study the effect of gene silencing in A. niger.
In this paper, we describe a method for simple construction of dsRNA-expression vectors under the control of a fungal constitutive promoter. This method makes use of homologous recombination between a general destination vector and a specific entry clone to generate the corresponding dsRNA expression vector (Gateway). The approach has the advantage of an easy and efficient exchange of DNA fragments to produce a dsRNA-expression vector. Using this approach, we were able to target for silencing the transcriptional activator XlnR in A. niger. We have analysed the effects of silencing of the transcriptional regulator towards the total expression of xylanases by functional assays and by measuring relative expression levels of two highly transcribed target genes encoding d-xylose reductase and endo-β-1,4-xylanase B.
Materials and methods
Fungal strains and media
A. niger strains used were derived from strain NW219 (cspA1 nicA1 leuA1 pyrA6), a derivative of CBS 120.49. Solid and liquid media were based on minimal medium (MM; Pontecorvo et al. 1953), contained 1 ml of trace elements solution (Vishniac and Santer 1957) per litre of medium and were supplemented with 16 μM nicotinic acid, 1.5 mM leucine and 5.0 mM uridine. For strains transformed by the pyrA-containing plasmid pGW635 (Goosen et al. 1989), uridine was not added to the culture medium.
Construction of destination and expression vectors
Vector pIM3710 (Benen et al. 1999) was double digested with NsiI and NotI, and a 3.6-kb fragment was isolated from agarose and blunt-ended by T4 DNA polymerase. Vector pK7GWIWG2(I) (Karimi et al. 2002) was partially digested with EcoRV, and a 4.1-kb fragment was isolated from agarose. The 4.1-kb fragment (with ccdB genes and homologous recombination sites in inverted repeat) was inserted by ligation into the 3.6-kb fragment of pIM3710, between the pyruvate kinase (pkiA) promoter and the endo-polygalacturonase II (pgaII) terminator from A. niger. Escherichia coli strain resistant to the toxic effects of ccdB gene product (One Shot® ccdB Survival™ T1 Phage Resistant, Invitrogen, Carlsbad, CA, USA) was used for propagation of the resulting destination vector pFIRD1. Transformation of One Shot® ccdB Survival™ T1 Phage-Resistant Cells with pFIRD1 was performed according to manufacturer’s instructions.
The expression vector pXLNRir for silencing of xlnR was made as follows. A fragment of 834 bp of xlnR coding region (plus a total of 58 bp for attB recombination sites) was amplified by polymerase chain reaction (PCR) using complementary DNA (cDNA; GenBank accession no. AJ001909) isolated from A. niger NW219 as template and primers attB1-xlnR and attB2-xlnR (Table 1).
The following steps were performed using Gateway technology (Invitrogen), according to manufacturer’s instructions. Polyethylene glycol (PEG)/MgCl2 purified attB-product and pDONR201 were mixed with BP clonase II in a 10-μl reaction mixture (BP reaction). Entry clone and destination vector pFIRD1 were mixed with LR clonase plus in a 20-μl reaction mixture (LR reaction). In both reactions, mixtures were incubated overnight at 25°C and treated with proteinase K. DH5α E. coli was electrotransformed by DNA from the reaction mixture. After overnight incubation at 37°C on Luria–Bertani (LB) kanamycin (50 μg∙ml−1) or ampicillin (100 μg∙ml−1) plates, colonies were picked, cultured on liquid LB medium with the same antibiotic composition and incubated at 37°C overnight. Entry clones and expression vectors for RNA-mediated silencing were isolated with the Wizard Plus Minipreps DNA Purification System (Promega, Madison, WI, USA).
Fungal transformation was performed as described elsewhere (Kusters-van Someren et al. 1991). In brief, enzymatic degradation of fungal cell wall was used to generate protoplasts, followed by purification by filtration and storage in isotonic sorbitol solution. For PEG-mediated transformation, protoplasts were incubated with 1 μg of pGW635 for selection and 30 μg of co-transforming pXLNRir for silencing of xlnR. Protoplasts transformed with 1 μg of pGW635 served as control for the transformation procedure. After this procedure, protoplasts were dispersed in sucrose-stabilised MM 0.6% (w/v) agar (0.95 M sucrose, pH 6.0) and added on sucrose-stabilised MM 1.2% (w/v) agar with identical composition. Plates were incubated 4 days at 30°C. After this period, spores were transferred from each strain to individual MM agar plates containing 50 mM glucose and supplements.
Semi-quantitative assays for endoxylanase activity and β-xylosidase/xylanase activities
Spores of each isolated A. niger strain (transformed by pGW635 only or by pGW635 and pXLNRir) were harvested from individual MM agar plates. Saline–Tween sterile solution [0.9% (w/v) NaCl, 0.005% (v/v) Tween-80] was used for harvesting spores. For screening on endoxylanase activity and on β-xylosidase/xylanase activities, 5 × 104 and 5 × 103 spores of each strain were dotted on plate. Endoxylanase activity was assayed as described elsewhere (Hasper et al. 2004) on MM agar plates with 25 mM d-xylose and 0.1% (w/v) azurine-dyed and cross-linked xylan [AZCL-xylan (Megazyme, Ireland)]. β-Xylosidase/xylanase activities were assayed on MM agar plates with 0.1% oat spelts xylan (Sigma Chemical, St Louis, MO, USA) and 0.5 mM 4-methylumbelliferyl-β-d-xylopyranoside [MUX (Sigma Chemical)]. Plates assayed for β-xylosidase/xylanase activities were incubated for 16 h at 30°C, and halo formation was detected under UV light (λ = 280 nm).
Transfer experiments and sample quenching
For transfer experiments, cultures were made in triplicate for each strain (transformed by pGW635 only or pGW635 and pXLNRir). Spores were inoculated at a final concentration of 106 spores∙per millilitre in MM containing 0.1% (w/v) yeast extract, 0.1% (w/v) casamino acids, and 100 mM d-fructose. Cultures were grown at 30°C in shake flasks in an orbital shaker at a constant agitation speed of 250 rpm for 18 h. Mycelia were subsequently harvested, transferred as described elsewhere (van Peij et al. 1998a) and grown for 4 h in MM with 50 mM d-xylose for induction. After this, mycelial samples were recovered by filtration on nylon gauze, quenched in liquid nitrogen and ground in a Mikro-dismembrator II (B. Braun, Melsungen, Germany). Frozen ground mycelial samples were stored at −80°C until further processing.
RNA isolation and cDNA synthesis
Frozen ground mycelial samples from transfer experiments were homogenised and lysed in Trizol Reagent (Invitrogen). Chloroform was added, and the mixtures were separated into aqueous and organic phases by phase-lock gel heavy (Eppendorf, Hamburg, Germany) according to manufacturer’s instructions. The aqueous phases were transferred to an identical volume of 70% (v/v) ethanol and loaded onto RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) columns for isolation and purification of RNA according to manufacturer’s instructions. RNA quantity and purity was determined by spectrophotometry in a NanoDrop-1000 instrument (NanoDrop Technologies, Wilmington, DE, USA). Mixtures of 1 ng of 1.2 kb kanamycin control RNA (Promega, San Luis Obispo, CA, USA) with 1 μg of isolated RNA were treated with 0.1 U∙μl−1 DNase I (amplification grade, Invitrogen), incubated at 37°C for 15 min, and DNase I was subsequently heat-inactivated according to manufacturer’s recommendations. The DNase-treated RNA mixtures were used as template for Reverse Transcription reactions with Omniscript RT kit (Qiagen), using 1.0 μM Oligo(dT)18, 0.5 mM each deoxyribonucleotide triphosphate, RT buffer and 0.2 U∙μl−1 Omniscript Reverse Transcriptase. The RT mixture was incubated for 1 h at 37°C, the resulting cDNA mixture was diluted in diethyl pyrocarbonate-treated water (1:20) and stored at −20°C. Each cDNA sample was synthesised from each biological replicate of three independent cultures.
Quantitative real-time PCR
Primers for cDNA of xlnR, xyrA, xynB (GenBank accession nos. AJ001909, AM269959, and AM269952) and the normaliser kanamycin external transcript (Table 1, under the label “qpcr”) were designed for specific melting temperatures (60°C ± 1°C), GC content (50% ± 5%) and amplicon sizes (139 to 150 bp), using the program Primer3. Reagent preparation and sample pipetting for real-time PCR were performed in the CAS-1200 (Corbett Life Science, Sydney, Australia) automated system. Reaction mixtures for real-time PCR contained 25% (v/v) diluted cDNA as template (dilution, 1:20), 0.15 μM each primer and ABsolute QPCR SYBR Green Mix (ABgene, Epsom, UK). Rotor-Gene 3000 (Corbett Life Science) was used for thermal cycling and real-time detection of DNA. Thermal cycling program included enzyme heat activation, amplification and melting steps. Two features of Rotor-Gene 6 software (Corbett Life Science) were used: melting analysis and comparative quantitation. The melting analysis feature was used to determine primer–dimer formation, and the comparative quantitation feature was used to determine take-off (TO) and amplification (A) values. The estimated values were used to calculate relative gene expression levels using the Pfaffl method (Pfaffl 2001), normalised to the added kanamycin external transcript (proportional to total RNA used for cDNA synthesis) and to a standard condition (normalised expression of control A. niger NW219 transformed by pGW635 only). From each synthesised cDNA, at least three runs were considered for analysis.
Genotyping by PCR
Frozen ground mycelial samples from transfer experiments were used for isolation of genomic DNA. Genomic DNA was isolated as described elsewhere (de Graaff et al. 1988). Starting with adequate primers (Table 1) and genomic DNA as template, PCR was used to evaluate three distinct aspects of the co-transformed strains: (1) integrity of native xlnR, (2) integrity of the silencing construct pXLNRir and (3) copy numbers of pXLNRir.
Integrity of native xlnR
PCR with primers xlnR-borderF and xlnR-borderR was used to detect native xlnR. These primers were designed to anneal upstream and downstream of the 834-bp region of xlnR targeted to silencing by pXLNRir, yielding a product of 874 bp.
Integrity of pXLNRir
Two regions of the silencing construct pXLNRir were targeted for PCR amplification: the upstream region spanning the pkiA promoter and part of the spacer sequence (primers used were PpkiA-spacer F and PpkiA-spacer R) and the downstream region spanning part of the spacer sequence and the pgaII terminator (primers used were Spacer-TpgaIIF and Spacer-TpgaIIR). In addition, a portion of inverted repeat of pXLNRir was targeted for amplification by single-primer PCR with primer IR-spacer.
Copy numbers of pXLNRir
Real-Time PCR was used to determine copy numbers of pXLNRir, with a primer pair specific for xlnR present in the inverted repeat and endogenous gene (xlnRir-copyF and xlnRir-copyR) and another pair for the normaliser gene pkiA (pkiA-copyF and pkiA-copyR). Reaction mixtures for real-time PCR contained about 10 ng∙μl−1 genomic DNA as template, 0.15 μM each primer and ABsolute QPCR SYBR Green Mix (ABgene, Epsom, UK). Thermal cycling conditions were as mentioned above for quantitative real-time PCR. For copy number determination, the Pfaffl method (Pfaffl 2001) was used: Samples were first normalised for genomic DNA quantity using the control pkiA gene in order to determine the total number of copies of xlnR segments per genome. Afterwards, the number of xlnR segments was subtracted by one unit (one endogenous xlnR gene per genome) and divided by two (assuming that inverted repeats are integrated “en bloc” and without suffering rearrangements) to obtain the average number of inverted repeats per genome of each strain.
Results
Effect of pXLNRir on xylan-degrading activities of A. niger
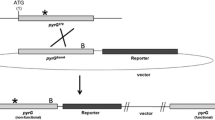
A. niger strain NW219 was transformed by pXLNRir (under the control of the constitutive pkiA promoter and pgaII terminator) and by pGW635, and strains were screened for xylan-degrading activities. The silencing vector pXLNRir was constructed from the reaction between the general destination vector pFIRD1 and an appropriate entry clone containing a portion of xlnR coding sequence (CDS) as depicted (Fig. 1). After transformation, 77 colonies were obtained with no visible differences in morphology or growth rate on complete medium (compared to non-transformed NW219 or NW219 transformed only with pGW635). These strains and one control strain (NW219::pGW635) were isolated and analysed for endoxylanase activity and β-xylosidase/xylanase activities. Under these conditions, a gradient of effects was observed in halo size and intensities of colour or fluorescence resulting from substrate degradation (Fig. 2). Compared to the control strain, nine strains (12% of 77) showed decreased activities in both assays, whereas three different strains showed decreased endoxylanase activity but no decreased β-xylosidase/xylanase activities, and one other strain showed decreased β-xylosidase/xylanase activities but no decreased endoxylanase activity. The nine strains that showed decreased activities in both assays were considered silenced transformant strains (either fully or partially silenced) and were further investigated, together with four non-silenced strains from co-transformation.

Construction of expression vectors for post-transcriptional gene silencing in A. niger. The arrow indicates the recombination reaction of pFIRD1 destination vector with two molecules of entry clone containing a partial coding sequence. ccdB-Cm R ccdB gene-chloramphenicol resistance gene; S spacer region

Semi-quantitative assays for analysis of xylan-degrading activities. a With AZCL xylan for detection of endoxylanase activity. b With MUX for detection of β-xylosidase and xylanase activities. C Wild-type strain (NW219::pGW635); numerals designate each silenced strain (NW219::pGW635::pXLNRir)
Expression of xlnR, xyrA and xynB in silenced strains
Quantitative real-time PCR was used to determine expression of xlnR, xyrA and xynB in the silenced strains and in four non-silenced strains relative to the control strain (NW219::pGW635). All samples were first normalised to the added kanamycin external transcript, directly proportional to the amount of total RNA. As a second normalisation step, gene expression relative to the control strain was calculated for each strain. Strain 50 was an outlier because, compared to the group of silenced strains, it showed significantly higher expression for all genes tested although still lower than the control strain (Fig. 3). In all silenced strains, relative gene expression was significantly decreased not only for the target gene xlnR but also for the regulated genes xyrA and xynB. Moreover, for each silenced strain, a moderate decrease in xlnR levels (to about 25–40% of wild-type expression levels) was associated with a larger decrease in xyrA and xynB expression levels (about 15% and 10% of wild-type expression levels, respectively). As expected, expression levels for xlnR, xyrA and xynB in the non-silenced strains were similar to wild-type levels within the experimental error.

mRNA levels for xlnR, xyrA and xynB in silenced and non-silenced strains, compared to wild type. C Wild-type strain (NW219::pGW635); numerals designate each strain from co-transformation (NW219::pGW635::pXLNRir). Error bars represent 95% confidence intervals (1.962 × SE)
Genome rearrangements and copy numbers of pXLNRir in silenced strains
Two approaches were taken for genotypic characterisation of all silenced strains: a qualitative approach based on conventional PCR and a more quantitative approach based on quantitative real-time PCR.
Through conventional PCR a fragment of about 874 bp of native xlnR was found in all strains using primers binding to xlnR but not to pXLNRir, indicating that, in each case, the endogenous xlnR locus was not affected (data not shown). Moreover, to detect genome rearrangements within integrated pXLNRir, two fragments covering the hairpin RNA expression cassette were amplified separately by PCR. Rearrangements were detected in all silenced strains (Fig. 4), although strains 6 and 30 mostly retained the two parts of the original construct according to this analysis. Strain 34 in contrast carried a truncated version of the hairpin RNA expression cassette at the 3′-region. The non-silenced strains also showed amplification of both fragments spanning the inverted repeat of pXLNRir (Fig. 4). PCR amplification of part of the inverted repeat region of pXLNRir followed by agarose gel electrophoresis showed that, within the group of silenced strains, a larger variety of fragments with stronger band intensity were produced compared to non-silenced strains (Fig. 5). Strain 6 was an outlier because an amplification profile more similar to that of non-silenced strains was obtained. In addition, specific amplification product was only obtained for strain 30. Furthermore, aspecific amplification was observed for the wild-type control strain (data not shown).

Genome integrity of pXLNRir. Genome integrity was assessed for both silenced (a and b) and non-silenced strains (c and d). a, c PCR-amplified 5′-region of pXLNRir. b, d PCR-amplified 3′-region of pXLNRir. Each triangle represents the expected fragment for intact pXLNRir. M Molecular weight marker; numerals designate each strain from co-transformation (NW219::pGW635::pXLNRir); C wild-type strain (NW219::pGW635)

Inverted repeats of pXLNRir. PCR-amplified portion of inverted repeat of pXLNRir from genomic DNA of non-silenced and silenced strains. Triangle Expected fragment for intact inverted repeat region. M Molecular weight marker; P silencing plasmid pXLNRir; C wild-type strain (NW219::pGW635); numerals designate each non-silenced and silenced strain (NW219::pGW635::pXLNRir)
For quantitative analysis, a portion of xlnR coding sequence present both in native xlnR and in pXLNRir was amplified and detected by real-time PCR. After normalization, diverse copy numbers were obtained, indicating variable frequencies of integration of pXLNRir in the different silenced strains (Table 2).
Discussion
Phenotypic screens are widely used for strain improvement in industry. For most industrial strains, a desirable phenotype such as increased or decreased enzyme production can be readily detected through simple functional assays. The system reported in this paper relies on downstream screening of easily characterisable phenotypes. Alternatively, in case a clear phenotypic difference cannot be obtained by plate screening of isolates, reverse-transcription PCR or Northern blot analysis may be used to confirm silencing. The Gateway-based technology presented in this study is based on exchange of fragments by means of two recombination reactions (BP and LR reactions). This recombination technology has the advantage that fragments originating from different genes can efficiently be inserted into an appropriate destination vector such as pFIRD1. For the construction of pXLNRir, a stem length of 834 nucleotides of xlnR coding sequence was used, in accordance to a previous finding that stem lengths of 600 or 900 nucleotides displayed higher silencing efficiencies in the ascomycete N. crassa (Goldoni et al. 2004).
After transformation of A. niger by pXLNRir, a gradient of effects was observed on xylan-degrading activities. Although the strains with decreased xylanolytic activity obtained presented a moderately efficient silencing degree (except for strain 50, about 30% the xlnR expression of wild-type A. niger), the number of silenced strains in comparison to the total number of strains obtained by co-transformation (12% of the total) was lower than expected. A number of reasons could serve as explanation for this result. First, some transformants might not be true co-transformants, even though this was not the case for the four non-silenced strains analysed. Second, some co-transformants such as non-silenced strain 54 suffered rearrangements in silencing construct. Third, silencing of xlnR was not significant in all cases to yield an obvious decrease of xylanolytic activities, such as in the case of non-silenced strains 3 and 54.
Two confounding factors in the analysis of RNAi transformants are genome rearrangements, which may occur after fungal transformation with silencing vectors and alternate silencing pathways not involving a typical Dicer-like/RISC-mediated degradation of mRNA (e.g. chromatin remodelling, DNA methylation and repeat induced mutation). In fact, non-silenced transformants often carry full silencing vectors, and silenced transformants often carry only altered versions of hairpin-expression units, as previously determined (Fitzgerald et al. 2004; Moriwaki et al. 2007; Nakayashiki et al. 2005). The separate analysis of either agarose gels of amplified regions of pXLNRir from genomic DNA or of estimated copy numbers of integrated pXLNRir is not conclusive. On the one hand, rearrangements may affect copy number determinations (even using Southern blotting techniques, which frequently miss smaller variations), and on the other hand, agarose gel analysis of amplified segments of genomic DNA is insufficient to provide reliable quantification. Nevertheless, combined analysis of these two methods may be used: Fr example, copy numbers of pXLNRir do not correlate with a higher degree of silencing efficiency, a situation similar to others reported (Nakayashiki et al. 2005; Nicolas et al. 2003; Tanguay et al. 2006; Fitzgerald et al. 2004; Henry et al. 2007; Yamada et al. 2007; Walti et al. 2006). Apart from genome instability and the occurrence of rearrangements, other factors may be responsible for a lower silencing efficiency of integrated hairpin-expression units. These factors include integration in regions of very low transcriptional activities (such as heterochromatin regions) or epigenetic modification of the inverted repeats without co-alteration of endogenous xlnR (possibly through RNA-induced transcriptional silencing). Due to the high genetic diversity and genome instability of strains transformed with dsRNA-expression vectors, it may be of advantage to pre-select silenced transformants for single-locus integration of silencing vector, as these transformants would be less likely to undergo genome rearrangements in successive generations.
In spite of the fact that strains were obtained with varying genotypes for integration of pXLNRir, it was possible to obtain a clear silencing effect both on enzyme xylan-degrading activities and on transcript levels of the genes xyrA (encoding d-xylose reductase) and xynB (also known as xlnB, encoding endo-β-1,4-xylanase B), previously shown to be positively regulated by XlnR in A. niger (Hasper et al. 2000).
In conclusion, the Gateway-based system presented in this paper has three noteworthy features: first, fast production of expression clones (about 1 week); second, use of any appropriate entry clone and third, no generation of unproductive clones. As shown for the transformation of the industrial microorganism A. niger with pXLNRir, it is furthermore possible to silence transcriptional regulators and screen for strains with the most desirable properties, depending on the effects of silencing on the expression of regulated genes.
References
Barnes SE, Alcocer MJC, Archer DB (2008) siRNA as a molecular tool for use in Aspergillus niger. Biotechnology Letters 30:885–890
Benen JA, Kester HC, Visser J (1999) Kinetic characterization of Aspergillus niger N400 endopolygalacturonases I, II and C. Eur J Biochem 259:577–585
Berns K, Hijmans EM, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M, Kerkhoven RM, Madiredjo M, Nijkamp W, Weigelt B, Agami R, Ge W, Cavet G, Linsley PS, Beijersbergen RL, Bernards R (2004) A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428:431–437
Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, Koch B, Haas SA, Paro R, Perrimon N (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303:832–835
Catalanotto C, Azzalin G, Macino G, Cogoni C (2000) Gene silencing in worms and fungi. Nature 404:245
Catalanotto C, Pallotta M, ReFalo P, Sachs MS, Vayssie L, Macino G, Cogoni C (2004) Redundancy of the two dicer genes in transgene-induced posttranscriptional gene silencing in Neurospora crassa. Mol Cell Biol 24:2536–2545
Cogoni C, Macino G (1997) Isolation of quelling-defective (qde) mutants impaired in posttranscriptional transgene-induced gene silencing in Neurospora crassa. Proc Natl Acad Sci U S A 94:10233–10238
Cogoni C, Macino G (1999a) Gene silencing in Neurospora crassa requires a protein homologous to RNA-dependent RNA polymerase. Nature 399:166–169
Cogoni C, Macino G (1999b) Posttranscriptional gene silencing in Neurospora by a RecQ DNA helicase. Science 286:2342–2344
de Graaff L, van den Broek H, Visser J (1988) Isolation and transformation of the pyruvate kinase gene of Aspergillus nidulans. Curr Genet 13:315–321
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811
Fitzgerald A, Van Kan JA, Plummer KM (2004) Simultaneous silencing of multiple genes in the apple scab fungus, Venturia inaequalis, by expression of RNA with chimeric inverted repeats. Fungal Genet Biol 41:963–971
Goldoni M, Azzalin G, Macino G, Cogoni C (2004) Efficient gene silencing by expression of double stranded RNA in Neurospora crassa. Fungal Genet Biol 41:1016–1024
Goosen T, van Engelenburg F, Debets F, Swart K, Bos K, van den Broek H (1989) Tryptophan auxotrophic mutants in Aspergillus niger: inactivation of the trpC gene by cotransformation mutagenesis. Mol Gen Genet 219:282–288
Hasper AA, Visser J, de Graaff LH (2000) The Aspergillus niger transcriptional activator XlnR, which is involved in the degradation of the polysaccharides xylan and cellulose, also regulates D-xylose reductase gene expression. Mol Microbiol 36:193–200
Hasper AA, Trindade LM, van der Veen D, van Ooyen AJ, de Graaff LH (2004) Functional analysis of the transcriptional activator XlnR from Aspergillus niger. Microbiology 150:1367–1375
Henry C, Mouyna I, Latge JP (2007) Testing the efficacy of RNA interference constructs in Aspergillus fumigatus. Curr Genet 51:277–284
Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J (2003) Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421:231–237
Karimi M, Inze D, Depicker A (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7:193–195
Kusters-van Someren MA, Harmsen JA, Kester HC, Visser J (1991) Structure of the Aspergillus niger pelA gene and its expression in Aspergillus niger and Aspergillus nidulans. Curr Genet 20:293–299
Meyer V (2008) Genetic engineering of filamentous fungi—progress, obstacles and future trends. Biotechnol Adv 26:177–185
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CA, Ram AF (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775
Moriwaki A, Ueno M, Arase S, Kihara J (2007) RNA-mediated gene silencing in the phytopathogenic fungus Bipolaris oryzae. FEMS Microbiology Lett 269:85–89
Mouyna I, Henry C, Doering TL, Latge JP (2004) Gene silencing with RNA interference in the human pathogenic fungus Aspergillus fumigatus. FEMS Microbiol Lett 237:317–324
Nakayashiki H, Hanada S, Nguyen BQ, Kadotani N, Tosa Y, Mayama S (2005) RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet Biol 42:275–283
Napoli C, Lemieux C, Jorgensen R (1990) Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2:279–289
Nicolas FE, Torres-Martinez S, Ruiz-Vazquez RM (2003) Two classes of small antisense RNAs in fungal RNA silencing triggered by non-integrative transgenes. Embo J 22:3983–3991
Paddison PJ, Silva JM, Conklin DS, Schlabach M, Li M, Aruleba S, Balija V, O’Shaughnessy A, Gnoj L, Scobie K, Chang K, Westbrook T, Cleary M, Sachidanandam R, McCombie WR, Elledge SJ, Hannon GJ (2004) A resource for large-scale RNA-interference-based screens in mammals. Nature 428:427–431
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JA, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EG, Debets AJ, Dekker P, van Dijck PW, van Dijk A, Dijkhuizen L, Driessen AJ, d’Enfert C, Geysens S, Goosen C, Groot GS, de Groot PW, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JP, van den Hondel CA, van der Heijden RT, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk PA, Lauber J, Lu X, van der Maarel MJ, Meulenberg R, Menke H, Mortimer MA, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NN, Ram AF, Rinas U, Roubos JA, Sagt CM, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJ, Wedler H, Wosten HA, Zeng AP, van Ooyen AJ, Visser J, Stam H (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25:221–231
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
Pontecorvo G, Roper JA, Hemmons LM, Macdonald KD, Bufton AW (1953) The genetics of Aspergillus nidulans. Adv Genet 5:141–238
Romano N, Macino G (1992) Quelling: transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol Microbiol 6:3343–3353
Ruijter GJ, Panneman H, Xu D, Visser J (2000) Properties of Aspergillus niger citrate synthase and effects of citA overexpression on citric acid production. FEMS Microbiol Lett 184:35–40
Tanguay P, Bozza S, Breuil C (2006) Assessing RNAi frequency and efficiency in Ophiostoma floccosum and O. piceae. Fungal Genet Biol 43:804–812
van der Krol AR, Mur LA, Beld M, Mol JN, Stuitje AR (1990) Flavonoid genes in petunia: addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell 2:291–299
van Peij NN, Gielkens MM, de Vries RP, Visser J, de Graaff LH (1998a) The transcriptional activator XlnR regulates both xylanolytic and endoglucanase gene expression in Aspergillus niger. Appl Environ Microbiol 64:3615–3619
van Peij NN, Visser J, de Graaff LH (1998b) Isolation and analysis of xlnR, encoding a transcriptional activator co-ordinating xylanolytic expression in Aspergillus niger. Mol Microbiol 27:131–142
Vishniac W, Santer M (1957) The thiobacilli. Bacteriol Rev 21:195–213
Walti MA, Villalba C, Buser RM, Grunler A, Aebi M, Kunzler M (2006) Targeted gene silencing in the model mushroom Coprinopsis cinerea (Coprinus cinereus) by expression of homologous hairpin RNAs. Eukaryot Cell 5:732–744
Yamada O, Ikeda R, Ohkita Y, Hayashi R, Sakamoto K, Akita O (2007) Gene silencing by RNA interference in the koji mold Aspergillus oryzae. Biosci Biotechnol Biochem 71:138–144
Acknowledgements
This research was funded by the Kluyver Centre for Genomics of Industrial Fermentation, which is supported by the Netherlands Genomics Initiative.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Oliveira, J.M., van der Veen, D., de Graaff, L.H. et al. Efficient cloning system for construction of gene silencing vectors in Aspergillus niger . Appl Microbiol Biotechnol 80, 917–924 (2008). https://doi.org/10.1007/s00253-008-1640-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-008-1640-x