
Abstract
Association mapping is considered to be an important alternative strategy for the identification of quantitative trait loci (QTL) as compared to traditional QTL mapping. A necessary prerequisite for association analysis to succeed is detailed information regarding hidden population structure and the extent of linkage disequilibrium. A collection of 430 tetraploid potato cultivars, comprising two association panels, has been analysed with 41 AFLP® and 53 SSR primer combinations yielding 3364 AFLP fragments and 653 microsatellite alleles, respectively. Polymorphism information content values and detected number of alleles for the SSRs studied illustrate that commercial potato germplasm seems to be equally diverse as Latin American landrace material. Genome-wide linkage disequilibrium (LD)—reported for the first time for tetraploid potato—was observed up to approximately 5 cM using r 2 higher than 0.1 as a criterion for significant LD. Within-group LD, however, stretched on average twice as far when compared to overall LD. A Bayesian approach, a distance-based hierarchical clustering approach as well as principal coordinate analysis were adopted to enquire into population structure. Groups differing in year of market release and market segment (starch, processing industry and fresh consumption) were repeatedly detected. The observation of LD up to 5 cM is promising because the required marker density is not likely to disable the possibilities for association mapping research in tetraploid potato. Population structure appeared to be weak, but strong enough to demand careful modelling of genetic relationships in subsequent marker-trait association analyses. There seems to be a good chance that linkage-based marker-trait associations can be identified at moderate marker densities.
Similar content being viewed by others


Introduction
Association mapping has become a customary approach to identify quantitative trait loci (QTL) responsible for variation in complex traits, complementary to traditional QTL mapping, because advances in molecular marker technology and statistical methods have made association mapping accessible and affordable, also to plant breeders (Zhu et al. 2008). Two major advantages inherent to association mapping are (1) a collection of variously related cultivars and breeding material includes all relevant allelic diversity and provides more generic results, and (2) a higher mapping resolution may be reached as more meiotic recombination events are sampled as compared to a bi-parental segregating mapping population (Flint-Garcia et al. 2003; Gaut and Long 2003; Jannink and Walsh 2002).
Specifically, such a germplasm collection can also impede the interpretation of the results because population structure and familial relationships among genotypes can negatively affect the outcome of association mapping studies by causing false positives (Flint-Garcia et al. 2003; Zhu et al. 2008). Therefore, a common strategy in association studies is to first inspect the germplasm collection for putative population structure followed by incorporation of correction factors for group effects when deemed necessary. The idea here is that only true associations—caused by physical linkage—will remain (Yu et al. 2006).
There are several ways to uncover population structure in a collection of cultivars and subsequently incorporate that information into association analysis. The software package Structure (Pritchard et al. 2000) assigns, within a Bayesian framework, group membership probabilities to each genotype using molecular marker information. Subsequently, marker-trait association analysis can take place within the identified groups (Remington et al. 2001; Simko et al. 2004b). Alternatively, the group membership probabilities can be translated into an extra set of covariables or a factor in a statistical model relating phenotypes to genotypes (Thornsberry et al. 2001). Another way to classify genotypes is based on standard multivariate analysis methods like clustering, where the input matrix of genetic distances can be derived from either molecular marker data or pedigree information. Identified groups from cluster analysis can subsequently be used as a factor in association analysis (Kraakman et al. 2004; Simko et al. 2004a). A more direct approach is to construct a genetic relatedness matrix, based on molecular marker or pedigree data, to impose structure on the variance–covariance matrix of the genetic effects (Malosetti et al. 2007; Parisseaux and Bernardo 2004). Yu et al. (2006) have included both a factor representing population structure and a genetic relatedness matrix in a mixed model framework for association analysis.
The feasibility of association mapping within a given species, i.e. the power to assess marker-trait associations, depends on the rate of linkage disequilibrium (LD) decay between loci, which relates to the number of meiotic generations since the most recent common ancestor (MRCA). A lower decay rate will support the detection of marker-trait associations with fewer markers, whereas faster LD decay will favour a better mapping resolution. For slowly decaying LD, whole-genome association scans become realistic (Breseghello and Sorrells 2006; Mackay and Powell 2007).
The cultivated potato, a predominantly vegetatively propagated autotetraploid crop species (2n = 4x = 48), typically represents a model species with few meiotic generations since its introduction in Europe. Potato offers opportunities to assess and compare methodologies for the detection of population structure and the estimation of LD decay, initially developed within a diploid context, at a higher ploidy level. Population structure and LD have previously been examined in potato within an association mapping context by Simko et al. (2004a, b), Gebhardt et al. (2004), Malosetti et al. (2007) and D’hoop et al. (2008). None of them have reported statistically significant population structure and LD decay estimates varied from rapidly decaying below a threshold of r 2 = 0.1 (<1 cM: Gebhardt et al. 2004) over a slower decay (~3 cM: D’hoop et al. 2008) to a long-range decay of about 10 cM (Simko et al. 2004a). Unfortunately all these estimates were based on a limited number of markers or just a localised attempt using a few DNA sequences. In contrast to association studies at the tetraploid level, a large number of QTL mapping studies have been performed at the diploid level (e.g., Costanzo et al. 2005; Malosetti et al. 2006; Werij et al. 2007), as well as some studies at the tetraploid level (Bradshaw et al. 2004, 2008; Khu et al. 2008). The value of association mapping in tetraploid potato, compared to conventional QTL mapping, resides in the agronomical relevance of the germplasm that is studied, and the relevant ploidy level where eventually marker-assisted selection is to be applied.
In this paper, we present evidence for population structure as detected in a large germplasm collection of tetraploid potato cultivars and progenitor clones, based on a substantial number of AFLP® and SSR markers. These results, as obtained with three methods: a Bayesian approach, a hierarchical clustering analysis and a factorial analysis (principal coordinate analysis), are compared and discussed. With the same marker information we analysed the LD pattern along the potato genome and some specific characteristics of the potato genome are discussed. The resulting information on population structure and LD decay will be deployed in a comprehensive association mapping study currently undertaken.
Materials and methods
Plant material
We collected a representative subset of worldwide available commercial potato germplasm containing 221 tetraploid potato cultivars and progenitor clones. For details about criteria used to compose this set we refer to D’hoop et al. (2008). This initial core set was expanded in a later stage. The parents of the SHxRH diploid mapping population (van Os et al. 2006) were added to enable marker positioning. To enlarge diversity coverage, 17 extra tetraploid potato cultivars were included. And to represent the current Dutch breeding germplasm pool, 190 advanced breeder’s clones were added. The material of in total 430 potato genotypes was kindly provided by five Dutch breeding companies and several genebanks (see Acknowledgements). An overview with background information regarding all 430 genotypes is available (Online Resource 1). Phenotypic trait data, mainly agro-morphological and quality-related traits, were collected by five Dutch breeding companies through consecutive years of clonal selection. Leaf material was harvested from greenhouse-grown and in vitro-grown genotypes, was frozen with liquid nitrogen and stored at −80°C until DNA extraction.
Molecular marker analysis
DNA extraction was according to van der Beek et al. (1992). DNA quality and concentration were visually examined using ethidiumbromide-stained 1% agarose gels.
AFLP
AFLP markers were generated according to Vos et al. (1995) using 26 well-known EcoRI/MseI and 15 well-known PstI/MseI primer combinations: E + AAC/M + AGG, E + AAC/M + CAC, E + AAC/M + CAG, E + AAC/M + CCA, E + AAC/M + CCT, E + AAC/M + CTC, E + AAC/M + CTG, E + AAG/M + ACC, E + AAG/M + AGC, E + AAG/M + CAC, E + AAG/M + CGA, E + ACA/M + CAC, E + ACA/M + CAG, E + ACA/M + CCT, E + ACA/M + CTG, E + ACC/M + AGT, E + ACC/M + CAT, E + ACC/M + CTT, E + ACT/M + CTC, E + AGA/M + ACC, E + AGA/M + CAG, E + AGA/M + CAT, E + AGA/M + CTC, E + AGT/M + CAG, E + ATG/M + CTA, E + ATG/M + CTC, P + AC/M + AAC, P + AC/M + ACT, P + AC/M + AGC, P + AC/M + AGG, P + AC/M + AGT, P + AC/M + ATG, P + AG/M + AAG, P + AG/M + ACC, P + AG/M + AGC, P + AG/M + AGT, P + AT/M + AAC, P + AT/M + AGG, P + CA/M + ACT, P + CA/M + AGG, P + CT/M + AGG. Fragments were separated using a capillary sequencer (MegaBACE 1000, Molecular Dynamics and Amersham, serial number 13757) according to van Eijk et al. (2004), each primer combination being labelled with either FAM, NED or JOE. The ROX channel was used for the MegaBACE™ ET900-ROX size standard from GE Healthcare (Amersham Biosciences). Pseudo gel images were scored with proprietary software at Keygene N.V. (Wageningen, NL). Marker nomenclature was based on the restriction enzyme combination, selective nucleotides and fragment mobility relative to a ROX-labelled size ladder.
Normalisation of signal intensity variation between capillaries due to DNA loading effects was performed by deducting genotype means (lane/column means) from the log-transformed band intensity values. Effectively, the residual log intensities were retained after fitting a genotype main effect to the genotype-by-marker log intensities, using the ANOVA procedure in GenStat, 11th edition (VSN International Ltd., Oxford, UK).
Information on the position of the AFLP markers was retrieved from the ultra-dense potato map, using the parental diploid genotypes SH83-92-488 and RH89-039-16 as internal reference (van Os et al. 2006; http://www.plantbreeding.wur.nl/potatomap/).
SSR
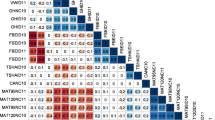
Fifty three microsatellite primer pairs (Table 1), previously designed based on expressed sequence tag database information (Bradshaw et al. 2008; Feingold et al. 2005; Ghislain et al. 2004; Kawchuk et al. 1996; Milbourne et al. 1998; Rios et al. 2007), were selected using the following criteria: (1) amplification products should map to a single locus (2) their quality score when available, (3) their linkage group and (4) their map location within a linkage group pursuing at least one SSR for each chromosome arm. Linkage group 8 is overrepresented in this set with 18 markers to be able to test our scoring methodology and linkage disequilibrium measure by deduction of genomic marker order from LD. Primer sequences, labelled with HEX, NED or 6-FAM, were modified by adding pigtail nucleotides according to Brownstein et al. (1996) to avoid as much as possible the appearance of stutter bands in the electropherograms. Microsatellites were amplified by separate PCR in a 20 μl reaction volume, containing 10 ng genomic DNA, 75 mM Tris–HCl pH 9.0, 20 mM (NH4)2SO4 0.1% (w/v) Tween 20, 2.5 mM MgCl2, 100 μM of each dNTP (Fermentas), 4 pmol of each primer and 0.3U Goldstar Taq DNA polymerase (Eurogentec). The optimised PCR conditions were one cycle of 94°C for 3 min, followed by 40 cycles of 94°C for 30 s, 50°C for 30 s, 72°C for 45 s and a final extension at 72°C for 10 min.
PCR amplification products were visually examined using ethidiumbromide stained 2% agarose gels along a 1-kb ladder (Invitrogen). Differently labelled PCR products (6-FAM, HEX and NED) were combined in appropriate amounts to obtain optimal peak patterns for detection. The fluorescently labelled products were separated by capillary electrophoresis using an ABI PRISM 3700 DNA Analyzer (Applied Biosystems). Electropherograms were created automatically using GeneScan Analysis Software v3.7 (Applied Biosystems). Peak mobilities and areas were determined using ABI PRISM Genotyper ® 3.6 NT software.
Allele calling was supported by peak mobility distribution plots because peak mobilities can vary within and between electrophoretic runs. Quantitative peak area information as provided by the Genotyper software was used to calculate pair-wise peak area ratios in order to determine allele copy numbers in individual samples according to the microsatellite allele counting-peak ratios (MAC-PR) methodology described in Esselink et al. (2004). Whenever four alleles were detected, single dosage was assumed for each allele as our samples were obtained from tetraploid individuals. When a single allele was detected, a dosage of four was suggested under the assumption of absence of null alleles. We do acknowledge the possibility of presence of non-detected null alleles as potato is highly heterogeneous. However, when no obvious proof of presence was available, absence of null alleles was maintained as valid genotypic model assumption. When unambiguous evidence of null alleles was available (e.g. a peak area ratio of ~2 or ~0.5 when only two alleles were detected or pair-wise peak area ratios of ~1 when only three alleles were detected), null alleles were defined and called as a separate allele “0” in the genotypic model as they represent an extra haplotype and so contain extra information. Also in this case we acknowledge that it is likely that obvious null alleles are not all of the same size and should therefore not be classified within one single allelic class. For convenience and to keep the statistical analysis simple we chose to lump all null alleles for a certain SSR locus into one single allelic class “0”.
Position information regarding the microsatellites was obtained from literature, or from the ultra dense potato map when polymorphic within the SHxRH population, using the parental diploid genotypes SH83-92-488 and RH89-039-16 as internal reference (van Os et al. 2006; http://www.plantbreeding.wur.nl/potatomap/).
As an approximate descriptive statistic for tetraploid potato, the Polymorphism Information Content (PIC) value was calculated according to Nei’s statistic (Nei 1973). Namely PIC = 1 − ∑p 2 i , where pi is the allele frequency, using co-dominant scores, of the ith allele of a certain microsatellite locus detected in the germplasm.
Population structure
Population structure in our diverse collection of potato cultivars was addressed using three different approaches. A clarifying overview is presented in Table 2, specifying the different marker sets, data types and analyses that have been used to assess population structure in our germplasm.
The first method is based on the Bayesian modelling environment implemented in the software Structure, v2.1 (Pritchard et al. 2000). This programme identifies putative subgroups based on the assumption of Hardy–Weinberg and absence of LD within subgroups. In that case LD originates principally from differences in allele frequencies between subpopulations. Based on position information and linkage configurations of the markers as obtained from the ultra-dense genetic map of potato (van Os et al. 2006), we created four different sets of polymorphic AFLP markers for the analysis with Structure. These four sets are (1) a set of 315 AFLPs, (2) a subset of 103 approximately independent and equidistantly spaced (every 5 cM) AFLPs, (3) a subset of 37 markers available from the 48 positions at the 12 centromeric marker clusters from both parental maps, linked in trans configuration and therefore haplotype specific and (4) a subset of 48 AFLPs from the 48 telomeric positions of the 12 chromosomes of both mapping parents. We will refer to these marker sets as the total mapped, equidistant, centromeric and telomeric set, respectively (Table 2). Structure was run under the assumption of admixture with independent allele frequencies. No a priori population information was used. Analyses were performed for the number of subgroups—K—ranging from two to 20 with two independent repeats for each K and with a total of 150,000 iterations of which the first 50,000 were considered as burn-in. Apart from this, Structure was also applied to the set of 53 microsatellites with the same assumptions and settings, although approximate mapping distance information was provided to Structure to account for positional clustering of some microsatellite loci. In all cases with AFLP data the ploidy was set to haploid, while with the SSRs ploidy was set to four with phase unknown.
The second method was a hierarchical clustering analysis performed with the programme DARwin (Dissimilarity Analysis and Representation for Windows v5.0.155, Perrier and Jacquemoud-Collet 2006). DARwin was run with four sets of AFLP data: (1) a set of 3,364 AFLP markers with normalised log-transformed band intensity information, (2) a subset of 1,772 polymorphic AFLPs using band presence/absence information, (3) a subset of 315 presence/absence polymorphic AFLPs with known map location and (4) an equidistantly spaced—approximately every 5 cM—subset of 103 AFLPs with presence/absence and band intensity information. We will refer to these marker sets as the complete, qualitative, total mapped and equidistant set, respectively. Aside from this, a set of 53 SSRs was used as well for clustering analysis with DARwin (Table 2). Due to the presence of null alleles, the genetic dissimilarities had to be calculated with GenStat 11th edition prior to data import in DARwin. For the calculation of the distance/(dis)similarity matrix, we opted for each analysis with presence/absence (AFLP) or allele dosage (SSR) data for the Jaccard similarity index (dissimilarity = 1 − similarity), whereas for the continuous AFLP band intensities we opted for an Euclidian distance-based dissimilarity index. For each dissimilarity calculation prior to clustering analysis with DARwin, 100 bootstraps were performed. For tree construction we opted for hierarchical clustering using the Ward minimal variance methodology. Information concerning market niche, year of registration, country of origin and group identity according to Structure was available as identifier set.
The third method was a factorial analysis, more specific a Principal Coordinate analysis (PCO), also realised with DARwin v5.0.155 (Perrier and Jacquemoud-Collet 2006). Factorial analysis was performed on two previously described sets of AFLP data: (1) the complete set and (2) the equidistant set using presence/absence data (Table 2). For both sets, the same dissimilarity matrices and identifier set were used as for the hierarchical clustering analysis.
In order to compare the Structure solution with the hierarchical clustering approach, a set of confusion matrices were composed, explicitly reporting misclassifications between both methods. Based on these confusion matrices values describing classification harmony could be calculated by dividing the sum of matches with the total sum of matches and mismatches (Story and Congalton 1986). To quantify how different cultivars classified into different groups according to Structure and DARwin actually are, an Analysis of Molecular Variance (AMOVA) was performed within the software package Arlequin v3.11 (Excoffier et al. 2005). For each AMOVA analysis the Euclidian distance matrix built with DARwin was used as input data. The number of permutations for AMOVA was set at 1,000. The fixation index (F ST), a measure used to quantify population differentiation calculated by Arlequin, was used for estimation of pair-wise differences between groups (Hudson et al. 1992).
Eigensoft 2.0 (Patterson et al. 2006; Price 2006) was engaged to perform a principal component analysis (PCA) with a selected set of 229 polymorphic AFLPs (a subset of the previously introduced total mapped set, where we excluded the monomorphic markers using presence/absence information) using presence/absence data and mapping information (Table 2). Marker data were normalised and principal components were tested for significance at the 0.001 threshold. Using the sum of squared loadings (normalised to unit length) of the significant principal components weighted by the square root of their Eigen value as a criterion, the most important markers were detected using GenStat 11th edition (VSN International Ltd., Oxford, UK). To identify which traits best support the groups that were detected previously, GenStat 11th edition was employed for a regression analysis using the genotypic main effects of individual phenotypic trait data that were estimated beforehand using appropriate mixed models, as response variables, and the coordinates of the two-first axes following PCO as explanatory variables. Only the trait best correlating with the variation in the PCO was maintained.
LD assessment
Linkage disequilibrium between loci was quantified with the squared correlation coefficient r 2 (Flint-Garcia et al. 2003; Zhao et al. 2005) between normalised log-transformed AFLP band intensities, see D’hoop et al. (2008) for a justification of the use of band intensities. Using a set of 720 AFLP markers with known map location within the ultra dense map (van Os et al. 2006), genome-wide LD was studied in potato. Provided with the information obtained through the population structure scan, LD decay was also examined within population groups. LD decay was visualised per chromosome by plotting r 2 versus map distance in centiMorgans.
The pattern of LD along the potato genome was also investigated for a set of 53 microsatellites, where we adapted the method of Flajoulot et al. (2005) for the calculation of r 2 based on microsatellite data to include full zygosity information on SSR alleles. As a measure for LD between two loci we first determined which allele was the most frequent one for each of the loci and then simply calculated the squared ordinary Pearson product moment correlation between the copy numbers, with possible values 0, 1, 2, 3, or 4, of these two most frequent alleles. Graphical representations of LD decay along the potato genome were accomplished with GenStat 11th edition (VSN International Ltd., Oxford, UK).
Results
Molecular marker analysis
A total of 3364 AFLP markers were collected from 41 AFLP fingerprints and analysed with proprietary software at Keygene (Wageningen, NL). The AFLP markers were studied in 430 tetraploid potato genotypes, including a threefold repetition of the diploid parents SH83-92-488 and RH89-039-16 of the ultra dense potato map (van Os et al. 2006) to assign marker names and positions to the AFLP markers. On average, one AFLP primer combination produced a fingerprint with 82 unambiguously distinguishable fragments. Of the 3,364 AFLP fragments, 628 did not display presence/absence segregation. Still, these “constant” bands may segregate as an allele dosage polymorphism, rendering predominantly quadruplex, triplex and perhaps some duplex genotypes, because allele frequencies higher than 0.75 suffice to cause phenotypically monomorphic bands. Of the remaining markers, 1,144 markers showed a clear presence/absence polymorphism based on intensity histograms and could therefore be scored in a dominant qualitative fashion. To allow us to use all 3,364 fragments for population structure and LD analyses, we worked with the quantitative band intensities (see D’hoop et al. (2008) for a justification of working with band intensities). This AFLP data set is comparable with scanning 23 Mb of DNA sequence for SNPs, because each AFLP fragment represents the scanning of 16 genomic nucleotides for SNP polymorphisms.
In total 720 markers, of which 315 were scored qualitatively, could be assigned to a genetic position on the ultra high density (UHD) potato map (van Os et al. 2006). We assumed that the position of these 720 markers in the diploid UHD map was essentially not different from the position of these bands in the collection of tetraploid genotypes. Although male and female cM positions may differ due to differences in recombination in male and female meiosis, we did not attempt to assign sex-averaged recombination distances between the markers. In Table 3 the distribution of the 720 mapped markers across the 12 potato chromosomes is quantitatively illustrated.
With 53 microsatellites or SSRs we could identify 653 alleles within the collection of 430 genotypes. On average 12 alleles per locus could be discerned, ranging from two for STM3020 to 26 for STM1100, while nearly two alleles were observed per tetraploid genotype. Given this large number of alleles, this results in an expansion of the number of possible genotypes in tetraploids. On average, each SSR locus had 75 different allelic combinations of which on average 37 were observed only once. Approximate Polymorphism Information Content (PIC) values based on allele frequencies ranged from 0.003 for STM3020 to 0.865 for STM5148, with an average of 0.663 (Table 1).
Population structure
The results obtained with Structure using the admixture model and assuming independent allele frequencies showed a continuous increase of the goodness of fit statistic, Ln[P(D)], versus the number of groups K. However, following the methodology presented by Evanno et al. (2005), we obtained a ΔK plot that clearly predicted the true underlying K (Online Resource 2).
Based on the ΔK plot, group characteristics and group membership probabilities, a six-group solution seemed the most adequate and is shown in Fig. 1. Fewer groups did not result in individual genotypes being allocated with high probability to particular groups, whereas a larger number of groups resulted in too high amounts of admixture affecting group membership probabilities. On the basis of our knowledge on individuals we named the groups in our six-group solution as “SH”, “Ancient”, “Processing”, “Starch”, “Fresh consumption” and “Rest” (Fig. 1). Three groups (“Starch”, “Processing” and “Ancient”) remained unchanged and also appeared at a lower number of hypothetical groups (K), but group members were swapped between repeats of these Structure runs.

Structure solution. Bar plot of individual potato cultivars generated by Structure 2.1 using the admixture model with independent allele frequencies. Marker data consisted of 103 AFLPs, spaced every 5 cM on the ultra dense potato genetic map (van Os et al. 2006). Groups are represented by colours, as indicated in the legend. Each column (430 in total) represents a cultivar its genotype and is partitioned into segments indicating its likely genetic origin. The longer a segment the more a genotype resembles one of the inferred six groups
The smallest group, “SH”, included merely the three repeated samples of the diploid mapping parent SH83-92-488. The “Ancient” group, the majority of which originated from the UK, comprised cultivars such as Paterson’s Victoria, King George, Sutton’s Flourball, Early Rose, etc., released between 1850 and 1950. The “Processing” group included cultivars related to Agria, a frying cultivar widely used as crossing parent, and therefore many modern breeds have Agria in their parentage. All cultivars belonging to the “Processing” group, using as criterion that membership probability exceeded 0.60, had an average underwater weight surpassing 400 g per 5 kg fresh weight. This value represents high dry matter content, typical for frying cultivars. The “Starch” group covered mostly those cultivars that were specifically bred for the starch industry. Their average underwater weight was 471 g per five kilogram fresh weight. The “Fresh consumption” group was the largest group with mainly European cultivars registered later than 1950 and intended for the fresh consumption market. “Fresh consumption” group members had an average underwater weight of 371 g per five kilogram fresh weight. The “Rest” group contained several progenitor clones, often used to introgress disease resistance, the diploid mapping parent RH89-039-16 and miscellaneous European cultivars.
With data from 53 microsatellite loci only two groups “Starch” and “Ancient” were stable across various K values, but low group membership probabilities for other genotypes did not allow confirmation of the grouping as obtained with AFLP data.
The second method to detect structure in potato germplasm was hierarchical clustering. The Ward tree, generated with DARwin software is shown in Fig. 2 (and Online Resources 3–6). In this tree the same six clusters of cultivars can be identified as obtained with Structure. Similarly as with the Structure analysis using SSR data, a Ward tree based on SSR data only recognised clusters similar to the groups “Starch” and “Ancient”.

Ward tree obtained with the complete set. The tree was created with DARwin 5 based on 3364 AFLP fragments using log-transformed normalised band intensities. Individuals have been given labels according to groups detected with Structure, restricted to group membership probabilities exceeding 0.7. The label undetermined (Und in the figure) refers to cultivars with group membership probabilities lower than 0.7
Principal coordinates analysis (PCO) was used as a third approach to detect population structure. Once again similar groups were identified as before with the Bayesian and hierarchical clustering analysis. The PCO plot using the complete set is presented in Fig. 3. The first two principal coordinates represent 5.91% of the total variation. In Online Resource 7 the PCO with the equidistant marker set using presence/absence data is depicted. Here the first two axes represented 7.71% of total variation. Visual inspection of both graphs showed that the PCO with the complete set demonstrated the best separation of groups of cultivars. Regression analysis of phenotypic data on the first two axes obtained through PCO revealed that the trait underwater weight was the best supporting trait for the separation between cultivars (Fig. 3). Squared correlation (adjusted r 2) amounted to 0.39, indicating that underwater weight could explain 39% of the structural variance depicted with the first two PCO axes. Following a principal components analysis (PCA) using a selected set of 229 polymorphic AFLPs with map location performed with Eigensoft, 15 principal components (PCs) appeared significantly associated with the represented molecular variance. The most discriminative AFLPs were identified by ranking the summed normalised loadings of all markers in each of these PCs (Table 4). Map location, summed normalised loading and associated potato quality traits based on previous association mapping results (D’hoop et al. 2008) are listed in Table 4.

Principal coordinate plot overlaid with the phenotypic trait best matching the variation based on regression analysis. The individuals are coloured with respect to their group identity according to Structure (70% group membership): green indicates starch, red indicates ancient, blue indicates fresh consumption, brown indicates processing cultivars and black represents SH. Light grey indicates undetermined cultivars (no group membership exceeding 0.7) together with the Rest group
Confusion matrices explicitly quantifying matches and mismatches between groups detected by Structure and DARwin were constructed. Table 5 presents an overview of the group harmony values that were calculated based on the constructed confusion matrices. A group harmony of 73.5% was found for the comparison between the five consistent groups obtained with Structure (thereby excluding the “Rest” group) and five visually distinguishable clusters acquired with DARwin using the complete set when the complete Structure solution was used, whereas a group harmony of 83.2% was found when the Structure solution was restricted to those cultivars having a group membership probability exceeding 0.70 (Table 5). When the comparison between Structure and DARwin using confusion matrices was limited to the solutions obtained with exactly the same marker data set—the equidistant set—group harmonies were lower when using band intensities: 65.4% when the entire Structure solution was concerned and 72.8% when comparison was limited to the cultivars with group membership probabilities higher than 0.70 (Table 5). However, when the same comparison was performed using presence/absence data group harmonies rose to 78.4 and 87.9%, respectively (Table 5). Through the equidistant set it was possible to explicitly test how well presence/absence data matched with normalised log-transformed band intensity data with respect to population structure. With Structure only presence/absence data could be used while with DARwin both normalised log-transformed band intensities and presence/absence data were examined. Correspondence between the Structure solution (“Rest” group excluded and restricted to ≥0.70 group membership probabilities) and hierarchical clustering with DARwin was 72.8 and 87.9% for the normalised log-transformed band intensities and presence/absence data, respectively (Table 5). Both group solutions obtained with Ward hierarchical clustering analysis within DARwin based on the same marker set corresponded to 58.5%. Matching improved to 67.6% when only cultivars belonging to a group (except the “Rest” group) with more than 0.70 probability according to Structure were considered (Table 5).
Analysis of molecular variance analyses were performed to quantify the differentiation between cultivar groups as identified by Structure (excluding the “Rest” group and restricted to cultivars with ≥70% group membership probability). Only 7 and 8.23% of the molecular variation could be attributed to the cultivar groups, using the complete or equidistant data set, respectively.
Assessment of linkage disequilibrium
AFLP markers were tested for pair-wise linkage disequilibrium by using the LD statistic r 2 (Flint-Garcia et al. 2003; Zhao et al. 2005). Only markers belonging to the same linkage group according to the genetic map of potato (van Os et al. 2006) were tested for LD. This resulted for each of the 12 linkage groups in an overview of LD decay, shown in Fig. 4. LD decay was estimated based on normalised log-transformed band intensities of 720 mapped AFLP markers. The general trend observed across the 12 linkage groups suggested that LD in potato decayed below 0.1—a threshold for significant LD that became widely accepted since its introduction by Kruglyak (1999) for human disease genetic data—when the genetic distance exceeded 5 cM. Yet, on several chromosomes secondary peaks of significant LD were visible beyond 5 cM (Fig. 4). This appeared to be an artefact due to the difference in the genetic positions of the centromeres of the respective parental linkage groups. The discrepancy between centromere positions is mainly caused by parent-specific differential recombination. The secondary peaks coincided exactly with the parental differences between centromeres; i.e. 15, 2, 5, 2, 2, 9, 2, 7, 8, 12, 11, and 5 cM for chromosomes 1 to 12, respectively. To confirm this explanation, markers were split according to whether they retrieved their genetic position in our reference map from the paternal or maternal linkage group. Subsequently separate LD decay plots were generated. This is illustrated for chromosome 1 in Fig. 5 (see Online Resources 08 to 18 for the other chromosomes). The secondary peak at 15 cM in Fig. 5 indeed disappeared, but some minor secondary peaks remained (e.g. at 20 cM). Those minor secondary peaks may equally well be due to AFLP marker clustering (cold spots for recombination) along haplotypes at a few cM distance.

Genome-wide LD in potato. LD decay across the 12 potato chromosomes based on 720 AFLP markers collected over 427 potato genotypes using log-transformed normalised band intensities. As LD measure r 2 has been used. Map positions in cM were deduced from the ultra dense potato genetic map (van Os et al. 2006). Each plot represents the LD pattern of one chromosome. The title of each plot mentions the number of markers between brackets that was used for the pattern reconstruction of a particular chromosome, e.g., 136 for chromosome 1

Parent-specific recombination and LD. Illustration of the effect of differential parental recombination on the LD decay of chromosome 1. On the left, the decay plot is shown for all 136 markers, in the middle the decay plot is shown for markers exclusively residing on the paternal map (RH, 45 AFLPs in total) and on the right the decay is presented for the 91 markers only residing on the maternal map (SH)
Apart from an overall per chromosome inspection, LD decay was examined within cultivar groups as well. This per group LD analysis was limited to the groups “Ancient”, “Fresh consumption”, “Processing” and “Starch” since these groups had an adequate number of cultivars with a higher than 0.70 group membership probability or could be identified by a relevant group characteristic. In Fig. 6 the LD decay plots of chromosome 1 are depicted for the four cultivar groups. Similar within-group LD decay plots can be found for the other potato chromosomes in Online Resources 19–29. LD patterns tremendously changed when studied within cultivar groups, and extended much farther. The inflation of LD greatly depended on the cultivar group, with the strongest effects for the Ancient and Starch groups. In Table 6 significance thresholds are presented for within-group LD for all chromosomes, based on the 0.95, 0.99 and 0.999 quantiles of the total distribution of r 2 for pair-wise marker combinations. For chromosome 1 the threshold for r 2 for significant LD using the 0.95 quantile for the “Ancient”, “Fresh consumption”, “Processing” and “Starch” group was estimated at r 2 = 0.27, 0.07, 0.10 and 0.16, respectively (Table 6). Based on these significance thresholds LD decayed on chromosome 1 within the “Ancient”, “Fresh consumption”, “Processing” and “Starch” group at about 15, 12, 13 and 13 cM, respectively. Still, in all four groups of chromosome 1 there were several pair-wise marker combinations with high r 2-values at distances beyond the LD decay border (Fig. 6).

Group-specific LD patterns for chromosome 1. From left to right LD decay plots are presented for four groups previously discovered with Structure and restricted to cultivars with group membership probabilities exceeding 0.7. For each plot, the title names first the group itself (Anc ancient; Fre fresh consumption; Pro processing; Sta starch) followed by the number of cultivars allocated to the group. In each case markers from both paternal and maternal map were combined, the total number of markers is mentioned in the title as well. Horizontal lines in each plot represent the calculated significance thresholds for the 0.95 quantile (striped) and the 0.99 quantile (full)
Linkage disequilibrium between 53 microsatellite markers as calculated with a modified statistic for r 2 (see M & M) resulted in an LD pattern along the potato genome. Because no common cM positions could be given since the microsatellites were mapped in different diploid mapping populations, the microsatellites were sorted according to their order on the chromosomes. It could be deduced from the pattern that a block of LD persisted on chromosome 8 (Online Resource 30). This was expected as part of the chromosome 8 microsatellites resided in or in the vicinity of the Granule Bound Starch Synthase (GBSS) locus. On chromosome 8 we deliberately elevated the number of SSR loci, even including multiple GBSS SSRs within a few kb physical distance, as a positive control to demonstrate that our LD statistic and scoring method for microsatellites performed as expected. This block of high LD values did not violate our previous estimation of LD decay based on AFLPs in potato as these SSRs were all localised within a 5-cM genetic interval. Because we selected microsatellite loci so distant that they presumably mapped at genetically independent loci, we did not expect to distinguish any other block of LD in this marker set, apart from the GBSS region on chromosome 8.
Discussion
A collection of tetraploid potato cultivars (Solanum tuberosum Group Tuberosum) representative for the cultivated gene pool in Europe and North America, was used to identify population structure and decay of linkage disequilibrium. The potato cultivars were analysed with two different marker methods because genome-wide SNP panels do not exist. First we will discuss the merits of the molecular data, second the population structure and third the decay of LD.
Although AFLP markers have several disadvantages when applied in diversity and association mapping studies in polyploids, such as their dominant inheritance and the risk of homoplasy (Woodhead et al. 2005; Zhu et al. 2008), the major advantage of AFLP fingerprinting of potato cultivars is the high multiplex ratio. This multiplex ratio of ~80 markers per assay results per data point in the most cost effective method, as compared to any other currently available system to detect genetic variation in potato (McGregor et al. 2000; Meudt and Clarke 2007). The high multiplex ratio also compensates the lack of co-dominance, because only four to ten times more dominant AFLP loci are required to obtain the same efficiency as with co-dominant markers (Mariette et al. 2002). Given the high heterozygosity of potato, more frequent low-informative marker loci are likely more efficient than less frequent but highly informative marker loci (Mariette et al. 2002).
In this study both qualitative presence/absence AFLP polymorphisms as well as quantitative normalised log-transformed band intensities were available, which allowed comparing results obtained with either data type. Where qualitatively scored AFLP data will have a certain error rate due to misclassification by the scoring software, the quantitative intensity values are not biased by these classification errors. In D’hoop et al. (2008) the authors explain that they opted for band intensities instead of presence/absence data for the estimation of LD and marker-trait associations because band intensity data were found related to allele dosage. Piepho (2001) has studied the issue in great detail and showed that the quantitative method will perform at least as good as presence/absence data, but he used diploids. In tetraploid potato, where zygosity of AFLPs can vary from one to four alleles, there is much more genetic information hidden in band intensities, that cannot be retrieved from a dominantly scored AFLP data set. This study illustrates the validity of these arguments in favour of using band intensities, because (1) the data set will contain more markers, including markers which could not be classified qualitatively as well as phenotypically monomorphic markers; (2) no major difference was observed with respect to the clusters obtained with DARwin; and (3) the correspondence between Structure and DARwin groupings improved (Table 5) when DARwin used the quantitative data set.
SSR
In this study 53 previously developed SSRs (Bradshaw et al. 2008; Feingold et al. 2005; Ghislain et al. 2004; Kawchuk et al. 1996; Milbourne et al. 1998; Rios et al. 2007) were used, with the intention to fully classify all four SSR alleles present in each of the 430 tetraploid potato cultivars.
To reach this goal microsatellite alleles were counted including the use of peak ratios (MAC-PR method; Esselink et al. 2004). This well-conceived method is nevertheless time-consuming and problematic due to preferential amplification of specific alleles, null-alleles, decreasing fluorescence signal intensity with increasing allele size or fade-out (Kimpton et al. 1993; Suenaga and Nakamura 2004). While counting alleles and converting the surface under the peak area into allele zygosity, these differential amplifications were taken into account, but we refrained from using allele-specific correction factors and did not seek to confirm null alleles. Instead, we assigned and scored null alleles when their presence was obvious. We do acknowledge that null alleles cannot be identified with this method when only one peak was detected (AAAA, AAA0 or AA00 etc.) or when two equal peaks were detected (AABB or AB00). Furthermore, the class of null alleles may comprise different null alleles, but technical homoplasy can also occur for normal alleles. Therefore, for convenience in subsequent statistical analyses all null alleles were lumped as one allele.
In total 653 alleles were detected with these 53 microsatellites, with an average of 12 alleles per locus. PIC values ranged from 0.450 to 0.865 (ignoring STM3020 and STM1054, Table 1). The low number of detected alleles for STM3020 and the low number of observed genotypes for both STM3020 and STM1054 (Table 1) strongly suggest a selective sweep or null alleles, because potato is a very heterozygous crop. Whereas PIC values are usually calculated on allelic phenotypes (ignoring zygosity of the alleles), we could calculate PIC values on the basis of full genotypic classification, which should result in slightly lower but more realistic values. In two previous microsatellite diversity studies in potato: one on 931 (Ghislain et al. 2004) and one on 742 (Ghislain et al. 2009) cultivated potato accessions including S. ajanhuiri, S. curtilobum, S. juzepczukii etc., PIC values ranged from 0.250 to 0.892. The PIC values of our study compare well with those observed by Ghislain et al. (2004, 2009), see Table 1. This is surprising, because our material from Group Tuberosum is only a narrow selection of the many cultivated potato species grown in Latin America. Even though we only intended to use PIC values in tetraploid potato for rough comparisons, this does indicate that the gene pool of tetraploid potato has not been narrowed due to commercial breeding efforts as sometimes suggested (Pavek and Corsini 2001). Furthermore, these results suggest that potato breeders are not likely to suffer from lack of genetic diversity in their future breeding efforts.
Population structure
Several sets of AFLP data differing either in their number of markers, their spacing or their data type and a set of microsatellites have been tested to ensure reliability and support for any inference made regarding population structure before embarking into association mapping analysis (Table 2). The Bayesian method (Structure) requires presence/absence data of independent marker loci. Subsets of 37 centromeric or 48 telomeric markers were selected to ensure marker independence. The ultra-dense potato genetic map (van Os et al. 2006) allowed us to guarantee that markers were only linked in repulsion. Telomeric positions on maternal or paternal linkage groups also guarantee independence. Unfortunately, these subsets did not offer sufficient statistical power due to their lower marker number. The equidistant marker set largely met the requirement of marker independence and produced a robust subdivision in six groups (Fig. 1) that made sense in view of the breeding history of potato. The past 150 years of potato breeding started with a population bottleneck due to the late blight epidemics in Europe (Irish Famine). The “Ancient” group resulted from cultivar by cultivar crosses. The desire to breed for pathogen resistance caused the development of progenitor clones with wild species introgression segments typically for the “Rest” group. Since several decades the commercial breeding companies focused on market niches, which seems to have caused further subdivision into the “Fresh consumption”, “Starch” and “Processing” group. The technical reasons to include experimental diploids as reference material resulted in a sixth group. The membership probabilities were at least 70%. Membership probabilities deteriorated when using the total mapped set of 315 AFLP markers with presence/absence data, probably due to lack of marker independence, because AFLP markers tend to cluster in the centromeric regions of chromosomes. The likelihood for this centromeric clustering increases when using more markers and when no control is imposed on the inter-marker distances, which is the case for the total mapped set compared to the equidistant set. In the series of Structure runs performed with the equidistant set, some of the groups already appeared at a stage as early as K equals three, and all the repeats at step K equals six revealed exactly the same group composition. Going beyond K equals six diffused the groups to such an extent that no interpretable group inference could be made anymore.
Additional evidence for the significance of the results obtained with Structure was provided by two DARwin analyses: (1) hierarchical clustering and (2) PCO. Besides the presence/absence data we could now use the complete set of 3364 AFLP markers as normalised log-transformed band intensities. Hierarchical clustering and PCO confirmed the “Starch”, “Processing”, “Fresh consumption”, “SH” and “Ancient” groups (Figs. 2+3, Online Resources 3 to 7).
Remarkably the SSR data were of little use to detect population structure. Both with Structure and DARwin only two consistent groups: “Ancient” and “Starch” were detected. Other groups previously detected with Structure and DARwin based on AFLP data could not be recovered with SSR as they were dispersed over several clusters.
Correspondence between Structure and DARwin results were quantified using confusion matrices (Table 5). Agreement values ranging from at least 65.4% to at most 87.9% confirm the relevance of the detected groups. Whenever the more stringent Structure solution—70% group membership—was used for comparison, a better correspondence was observed. This suggests that group membership probabilities should be considered when interpreting Structure results; a meaningful threshold for allocations of individuals to groups appeared to be 70%. Another apparent trend was that correspondence between the presence/absence data solution and the log-transformed band intensity solution increased when the latter was based on a larger data set. This is most likely due to band intensity being intrinsically more variable than presence/absence: intensities are continuous data whereas presence/absence data can in principle only take two values.
Earlier molecular marker studies on cultivated potato germplasm were unable to show significant population structure. Simko et al. (2004b) inspected without success a set of 150 tetraploid North American potato cultivars for population structure using segregating bands from 27 ISSR and RAPD markers. In another study, Simko et al. (2006) investigated 66 DNA fragments from 47 accessions for evidence of population structure using the Bayesian approach and again reported no significant overall substructure, but did report that individual fragments in proximity of an introgression locus for resistance did reveal subgroup presence. Gebhardt et al. (2004) suggested that the number of meiotic generations is insufficient to allow a genetic separation between cultivars, as well as the high familial relatedness between cultivars, when a set of 600 potato cultivars was analysed without providing evidence for population structure.
Our AMOVA analysis illustrates why others may not have been able to demonstrate population subdivision, because only 7% of the genetic variation can be assigned to our six groups. In addition, we did observe with Structure a continuously increasing goodness of fit (Ln[P(D)]) with an increasing number of (K) groups. In our opinion this suggests that the identification of a meaningful group structure could be based on specific regions in the genome, whereas the majority of the genome is still homogeneous with respect to the six groups. Introgression of wild species resistance genes could be the origin of the non-homogeneous regions in the potato genome. These findings suggest that to be able to perform reliable association mapping, i.e. with appropriate control for false positives, it will be of utmost importance to account for relationships—both obvious and subtle—between genotypes. Straightforward inclusion of structure groups as covariables in association models following their detection by Structure or DARwin will not suffice as they only describe obvious genotypic relationships. Imposing genetic relationship structure on the variance–covariance matrix of the genotypic effects in association mapping models using available marker information to fully account for both obvious and subtle genotypic relationships is probably the most adequate approach to preclude spurious associations (Malosetti et al. 2007; Yu et al. 2006).
In conclusion, the Bayesian, hierarchical clustering as well as the factorial approach to population structure discovery indicated that (1) the gene pool used to breed cultivars for starch industry differs significantly from other germplasm, (2) recent material diverges from older more alike potato germplasm. The current gene pool has expanded due to introgression of disease resistance from wild relatives and (3) there is no difference between germplasm pools used by different breeding companies aiming for the markets fresh consumption and processing.
Linkage disequilibrium
Detailed information concerning the decay of LD within the potato genome is presented. We used the squared correlation coefficient r 2 as LD measure, as proposed by Flint-Garcia et al. (2003) and Zhao et al. (2005). We found LD across population groups to decay on average at about 5 cM throughout the potato genome (Fig. 4), when applying a 0.1 cut-off value for detection of LD as proposed by Kruglyak (1999). Although microsatellite data may offer better resolution in terms of alleles, similar LD patterns were observed (Online Resource 30) with no LD extending beyond 5 cM.
Previous LD estimates in potato used data from either highly localised studies (Gebhardt et al. 2004) or pyrosequencing of 66 DNA fragments from only 47 accessions (Simko et al. 2006). Gebhardt et al. (2004) examined four markers within 1 cM on chromosome 5: for two markers residing within 0.3 cM LD was maintained, whereas for markers being separated by 0.6 cM LD had decreased and for markers being 0.9 cM apart linkage equilibrium had been reached. In our opinion this study severely underestimates LD, because the region is known to be a hotspot for recombination. Simko et al. (2006) concluded from their data that LD decayed below 0.1 at about 10 cM, but their DNA fragments were end-sequences of BACs containing R-genes from wild species. This may lead to an overestimation of LD due to the few meioses since introgression of R-genes.
Maize is an outbreeder like potato, where LD drops below 0.1 within 2kbp according to Remington et al. (2001). Comparison with the observed LD pattern across population groups of potato—LD below 0.1 at 5 cM, i.e., approximately 5,300 kbp considering the physical genome length of ~850 Mb for a genetic map length of ~800 cM (Bennett and Leitch 2004; Marie and Brown 1993; van Os et al. 2006)—proves that LD is maintained over a much larger distance within potato. This is most likely the result of the obligatory sexual reproduction in maize versus the clonal selection used in potato breeding, reducing the number of meioses considerably. Self pollinating crops—as opposed to outbreeders—usually display LD over much larger distances as a consequence of their mating system. In rice for example LD extends up to more than 60 cM before decreasing to 0.1 (Agrama et al. 2007) and in barley LD remains above 0.1 up to about 40 cM (Malysheva-Otto et al. 2006). As expected, potato demonstrates a by far higher decay rate. Sugarcane, a polyploid crop with a far more intricate genome than potato but exposing a similar breeding history and strategy, features a sharp drop in LD around 5 cM and a steady decrease towards 30 cM (Raboin et al. 2008). As compared to tomato, another solanaceous crop, LD within potato declines faster: significant LD for tomato (P value < 0.05 which corresponds to an r 2 of 0.37) was found between loci up to 20 cM apart (van Berloo et al. 2008).
As a measure for LD between two microsatellite loci, we adapted the method of Flajoulot et al. (2005) to calculate r 2 based on the most frequent allele to enable the use of zygosity information. Flajoulot et al. (2005) tested their LD measure using pairs of SSR loci on different chromosomes and illustrated that their LD measure was not likely to detect LD between unlinked loci. Our method also did not discover LD between loci on different chromosomes, and it did detect LD between closely mapped SSR loci, as expected.
A remarkable LD pattern can be distinguished on several chromosomes in Fig. 4: there seems to be significant linkage disequilibrium at specific positions beyond 5 cM. This is an artefact caused by position information of the AFLP markers used in this study. The position information is retrieved from the two parental maps of the ultra-dense potato map (van Os et al. 2006). Maternal or paternal centromeric marker dense clusters have been assigned to different cM positions, due to parental hot spots and cold spots for recombination. In spite of their localisation on independent haplotypes from either a maternal or paternal centromeric cluster, strong LD values have been observed between such centromeric markers. This artefact will disappear if sex-averaged map positions or physical map positions were known. When the marker data are split in parent-specific data sets, the artefact is also absent (Fig. 5 and Online Resources 08 to 18). The remaining parent-specific pair-wise marker pairs exhibiting higher LD beyond 5 cM reflect the presence of large haplotype blocks in the potato genome.
Considering that actual association analysis takes place within detected groups when correcting for population structure, it was interesting to zoom in on the LD pattern within the relevant observed groups found with Structure. LD within groups appeared much stronger when compared to the overall LD pattern. Chromosomal regions showing significant LD extended up to 15 cM for chromosome 1 (Fig. 6). In view of the prolonged LD (10 cM within groups) and a genome length of 800 cM, we expect that association studies can be performed with modest numbers of markers.
Long stretches of LD within detected groups have contrasting implications. On the one hand it is clear that association mapping may not result in fine mapping, but on the other hand detection of QTL using association mapping should be relatively straightforward—disregarding the complex nature of the potato genome.
The increased level of LD observed within specific cultivar groups—the “Starch”, “Processing” and “Fresh consumption” cultivars—confirms the relevance of our observation on population subdivision.
A highly relevant quality trait referring to dry matter or starch content was recorded in this study as “underwater weight” This phenotypic trait best corresponded with population structure (Fig. 3). It is clear that strong selection for underwater weight and presumably other yield-related traits, along with relaxed selection for quality traits in starch potatoes, have played a leading role in shaping today’s commercial potato germplasm. Additionally, underwater weight came forward as one of the traits with which the majority of markers, best explaining the molecular variation represented by the significant principal components according to PCA analysis, were associated (Table 4). The other traits associated with these most discriminative markers: after baking darkening, after cooking darkening and chipping colour, reflect to some extent that processing quality was another important aspect that helped creating the present day commercial potato germplasm. Further, it was striking to notice that the “Ancient” group always reflected the highest within-group LD when compared to the other groups (Fig. 6 and Online Resources 19 to 29). This provides some evidence for higher relatedness or alikeness of ancient cultivars when compared to more recent breeding material: most probably the result of the exploitation of wild germplasm within post-1950 breeding programmes.
Prospects/implications
We used various ways to investigate population structure and although there was no strong population structure, the different ways of identifying groups of genotypes coincided reasonably. We chose a pragmatic, heuristic approach to the identification of population structure, thereby sometimes ignoring the typicalities and complexities of tetraploid inheritance. For example, we applied the Bayesian clustering implemented in the Structure package to the binary AFLP data, where we told Structure to treat the potato genotypes as if they were haploid. This choice seems contradictory to the tetraploid nature of potato. In Structure the creation of groups is based on assigning genotypes to groups such that within a group genotype frequencies at individual loci follow from allele frequencies, while joint genotype frequencies at pairs of loci follow from genotype frequencies at individual loci. By choosing potato to be haploid, for AFLP, we merely tell Structure to abstain from looking at intra locus disequilibrium (absence of Hardy–Weinberg equilibrium) and concentrate on between locus disequilibrium (LD), where we admit to have worked with marker phenotypes (band presence/absence) instead of more informative marker genotypes.
Based on our investigation of the genetic diversity in our germplasm, we can conclude that for association mapping some form of correction for population stratification will be necessary to arrive at linkage-based associations between phenotypic trait loci and genotypic marker loci. As the population structure is weak, simple inclusion of a covariable or cofactor based on Structure group results will not suffice to preclude false positives. A more subtle correction will be necessary, by imposing relationship structure on the variance/covariance matrix of the genotypic effects, where the relations may be estimated from the complete set of markers (Yu et al. 2006). This imposition of a relationship matrix in an association mapping analysis will correct for false positive marker-trait associations (Malosetti et al. 2007). We anticipate that this approach will enable detection of valuable and reliable associated markers that may be useful for marker assisted breeding in potato using modest marker numbers.
References
Agrama H, Eizenga G, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breed 19:341–356
Bennett M, Leitch I (2004) Angiosperm DNA C-values database (release 5.0, Dec. 2004). http://www.kew.org/cvalues/homepage.html
Bradshaw JE, Pande B, Bryan GJ, Hackett CA, McLean K, Stewart HE, Waugh R (2004) Interval mapping of quantitative trait loci for resistance to late blight [Phytophthora infestans (Mont.) de Bary], height and maturity in a tetraploid population of potato (Solanum tuberosum subsp tuberosum). Genetics 168:983–995
Bradshaw JE, Hackett CA, Pande B, Waugh R, Bryan GJ (2008) QTL mapping of yield, agronomic and quality traits in tetraploid potato (Solanum tuberosum subsp tuberosum). Theor Appl Genet 116:193–211
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Brownstein MJ, Carpten JD, Smith JR (1996) Modulation of non-templated nucleotide addition by Taq DNA polymerase: primer modifications that facilitate genotyping. Biotechniques 20:1004–1010
Costanzo S, Simko I, Christ BJ, Haynes KG (2005) QTL analysis of late blight resistance in a diploid potato family of Solanum phureja × S. stenotomum. Theor Appl Genet 111:609–617
D’hoop B, Paulo M, Mank R, van Eck H, van Eeuwijk F (2008) Association mapping of quality traits in potato (Solanum tuberosum L.). Euphytica 161:47–60
Esselink GD, Nybom H, Vosman B (2004) Assignment of allelic configuration in polyploids using the MAC-PR (microsatellite DNA allele counting-peak ratios) method. Theor Appl Genet 109:402–408
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Feingold S, Lloyd J, Norero N, Bonierbale M, Lorenzen J (2005) Mapping and characterization of new EST-derived microsatellites for potato (Solanum tuberosum L.). Theor Appl Genet 111:456–466
Flajoulot S, Ronfort J, Baudouin P, Barre P, Huguet T, Huyghe C, Julier B (2005) Genetic diversity among alfalfa (Medicago sativa) cultivars coming from a breeding program, using SSR markers. Theor Appl Genet 111:1420–1429
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Gaut BS, Long AD (2003) The lowdown on linkage disequilibrium. Plant Cell 15:1502–1506
Gebhardt C, Ballvora A, Walkemeier B, Oberhagemann P, Schuler K (2004) Assessing genetic potential in germplasm collections of crop plants by marker-trait association: a case study for potatoes with quantitative variation of resistance to late blight and maturity type. Mol Breed 13:93–102
Ghislain M, Spooner DM, Rodriguez F, Villamon F, Nunez J, Vasquez C, Waugh R, Bonierbale M (2004) Selection of highly informative and user-friendly microsatellites (SSRs) for genotyping of cultivated potato. Theor Appl Genet 108:881–890
Ghislain M, Nunez J, del Rosario Herrera M, Pignataro J, Guzman F, Bonierbale M, Spooner DM (2009) Robust and highly informative microsatellite-based genetic identity kit for potato. Mol Breed 23:377–388
Hudson RR, Slatkin M, Maddison WP (1992) Estimation of levels of gene flow from DNA sequence data. Genetics 132:583–589
Jannink J-L, Walsh B (2002) Association mapping in plant populations. In: Kang MS (ed) Quantitative genetics, genomics and plant breeding. CAB International Publishing, Wallingford, pp 59–68
Kawchuk LM, Lynch DR, Thomas J, Penner B, Sillito D, Kulcsar F (1996) Characterization of Solanum tuberosum simple sequence repeats and application to potato cultivar identification. Am Potato J 73:325–335
Khu DM, Lorenzen J, Hackett CA, Love SL (2008) Interval mapping of quantitative trait loci for corky ringspot disease resistance in a tetraploid population of potato (Solanum tuberosum subsp tuberosum). Am J Potato Res 85:129–139
Kimpton CP, Gill P, Walton A, Urquhart A, Millican ES, Adams M (1993) Automated DNA profiling employing multiplex amplification of short tandem repeat loci. Genome Res 3:13–22
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet 22:139–144
Mackay I, Powell W (2007) Methods for linkage disequilibrium mapping in crops. Trends Plant Sci 12:57–63
Malosetti M, Visser RGF, Celis-Gamboa C, van Eeuwijk FA (2006) QTL methodology for response curves on the basis of non-linear mixed models, with an illustration to senescence in potato. Theor Appl Genet 113:288–300
Malosetti M, van der Linden CG, Vosman B, van Eeuwijk FA (2007) A mixed-model approach to association mapping using pedigree information with an illustration of resistance to Phytophthora infestans in potato. Genetics 175:879–889
Malysheva-Otto L, Ganal M, Roder M (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:6
Marie D, Brown SC (1993) A cytometric exercise in plant DNA histograms, with 2C values for 70 species. Biol Cell 78:41–51
Mariette S, Le Corre V, Austerlitz F, Kremer A (2002) Sampling within the genome for measuring within-population diversity: trade-offs between markers. Mol Ecol 11:1145–1156
McGregor CE, Lambert CA, Greyling MM, Louw JH, Warnich L (2000) A comparative assessment of DNA fingerprinting techniques (RAPD, ISSR, AFLP and SSR) in tetraploid potato (Solanum tuberosum L.) germplasm. Euphytica 113:135–144
Meudt HM, Clarke AC (2007) Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci 12:106–117
Milbourne D, Meyer RC, Collins AJ, Ramsay LD, Gebhardt C, Waugh R (1998) Isolation, characterisation and mapping of simple sequence repeat loci in potato. Mol Gen Genet 259:233–245
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Parisseaux B, Bernardo R (2004) In silico mapping of quantitative trait loci in maize. Theor Appl Genet 109:508–514
Patterson N, Price AL, Reich D (2006) Population structure and eigenanalysis. PLoS Genet 2:e190
Pavek J, Corsini D (2001) Utilization of potato genetic resources in variety development. Am J Potato Res 78:433–441
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. http://darwinciradfr/darwin
Piepho H-P (2001) Exploiting quantitative information in the analysis of dominant markers. Theor Appl Genet 103:462–468
Price AL (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Raboin L-M, Pauquet J, Butterfield M, D’Hont A, Glaszmann J-C (2008) Analysis of genome-wide linkage disequilibrium in the highly polyploid sugarcane. Theor Appl Genet 116:701–714
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Rios D, Ghislain M, Rodriguez F, Spooner DM (2007) What is the origin of the European potato? Evidence from Canary island landraces. Crop Sci 47:1271–1280
Simko I, Costanzo S, Haynes KG, Christ BJ, Jones RW (2004a) Linkage disequilibrium mapping of a Verticillium dahliae resistance quantitative trait locus in tetraploid potato (Solanum tuberosum) through a candidate gene approach. Theor Appl Genet 108:217–224
Simko I, Haynes KG, Ewing EE, Costanzo S, Christ BJ, Jones RW (2004b) Mapping genes for resistance to Verticillium albo-atrum in tetraploid and diploid potato populations using haplotype association tests and genetic linkage analysis. Mol Genet Genomics 271:522–531
Simko I, Haynes KG, Jones RW (2006) Assessment of linkage disequilibrium in potato genome with single nucleotide polymorphism markers. Genetics 173:2237–2245
Story M, Congalton RG (1986) Accuracy assessment: a user’s perspective. Photogramm Eng Remote Sens 52:397–399
Suenaga E, Nakamura H (2004) Rapid individual identification by minisatellite variant repeat (MVR)-PCR at D1S8 locus using exponential law. Anal Sci 20:1629–1633
Thornsberry JM, Goodman MM, Doebley J, Kresovich S, Nielsen D, Buckler ES (2001) Dwarf8 polymorphisms associate with variation in flowering time. Nat Genet 28:286–289
van Berloo R, Zhu A, Ursem R, Verbakel H, Gort G, van Eeuwijk F (2008) Diversity and linkage disequilibrium analysis within a selected set of cultivated tomatoes. Theor Appl Genet 117:89–101
van der Beek JG, Verkerk R, Zabel P, Lindhout P (1992) Mapping strategy for resistance genes in tomato based on RFLPs between cultivars: Cf9 (resistance to Cladosporium fulvum) on chromosome 1. Theor Appl Genet 84:106–112
van Eijk MJ, Broekhof JL, van der Poel HJ, Hogers RC, Schneiders H, Kamerbeek J, Verstege E, van Aart JW, Geerlings H, Buntjer JB, van Oeveren AJ, Vos P (2004) SNPWave: a flexible multiplexed SNP genotyping technology. Nucleic Acids Res 32:e47
Van Os H, Andrzejewski S, Bakker E, Barrena I, Bryan GJ, Caromel B, Ghareeb B, Isidore E, de Jong W, van Koert P, Lefebvre V, Milbourne D, Ritter E, Rouppe van der Voort JNAM, Rousselle-Bourgeois F, van Vliet J, Waugh R, Visser RGF, Bakker J, van Eck HJ (2006) Construction of a 10,000-marker ultra-dense genetic recombination map of potato: providing a framework for accelerated gene isolation and a genome-wide physical map. Genetics 173:1075–1087
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Werij JS, Kloosterman B, Celis-Gamboa C, de Vos CHR, America T, Visser RGF, Bachem CWB (2007) Unravelling enzymatic discoloration in potato through a combined approach of candidate genes, QTL, and expression analysis. Theor Appl Genet 115:245–252
Woodhead M, Russell J, Squirrell J, Hollingsworth PM, Mackenzie K, Gibby M, Powell W (2005) Comparative analysis of population genetic structure in Athyrium distentifolium (Pteridophyta) using AFLPs and SSRs from anonymous and transcribed gene regions. Mol Ecol 14:1681–1695
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhao H, Nettleton D, Soller M, Dekkers JCM (2005) Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet Res 86:77–87
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome 1:5–20
Acknowledgments
We express our gratitude to the potato breeding companies and gene banks that were willing to provide us with tubers and phenotypic data of potato cultivars. It concerns Agrico Research (Netherlands), Averis seeds (Netherlands), C. Meijer (Netherlands), HZPC Holland BV (Netherlands) and Van Rijn-KWS (Netherlands), Agriculture and Agri-Food (Canada), Arche Noah (Austria), IPK (Germany), INRA (France), SASA (Scotland), Teagasc (Ireland) and USDA (USA). Our special thanks go to Paul L.C. Keizer for his artistic creations within GenStat 11 and to Caroline Marquez-Castro for providing PCA results. The help of Brigitte Uwimana and Hanneke van der Schoot in SSR data generation is equally appreciated. We would also like to thank three anonymous reviewers for their valuable comments. This research project was financed by the Centre for BioSystems Genomics (CBSG) which is part of the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G. Bryan.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
Background information regarding the 430 cultivars involved in our study. Struc 70% refers to group identity according to STRUCTURE restricting to group membership probabilities of 0.7 or higher (XLS 97 kb)
Online Resource 2
Delta K plot illustrating the predicted number of groups with STRUCTURE (PDF 21 kb)
Online Resource 3
Ward tree obtained with the qualitative set. The tree was constructed with DARWIN 5 based on 1772 AFLP fragments using presence/absence data. Individuals received labels according to their group identity as detected with STRUCTURE, restricted to group membership probabilities exceeding 0.7. Colour codes are explained in the accompanying figure title (TIFF 4079 kb)
Online Resource 4
Ward tree deploying the total mapped set. The tree was built with DARWIN 5 based on presence/absence information on 315 AFLP markers. Individuals received labels according to their group identity as detected with STRUCTURE, restricted to group membership probabilities exceeding 0.7. Colour codes are explained in the accompanying figure title (TIFF 3805 kb)
Online Resource 5
Resulting Ward tree of the equidistant set using presence/absence data. The tree was made with DARWIN 5 using presence/absence information on 103 AFLP fragments. Individuals received labels according to their group identity as detected with STRUCTURE, restricted to group membership probabilities exceeding 0.7. Colour codes are explained in the accompanying figure title (TIFF 4942 kb)
Online Resource 6
Ward tree acquired with the equidistant set using band intensity data. The tree was composed with DARWIN 5 using normalised log-transformed band intensities of 103 AFLP fragments. Individuals received labels according to their group identity as detected with STRUCTURE, restricted to group membership probabilities exceeding 0.7. Colour codes are explained in the accompanying figure title (TIFF 3488 kb)
Online Resource 7
Factorial analysis realised with the equidistant AFLP set. The Principal Coordinate analysis was performed with DARWIN 5 using presence/absence data of 103 AFLP fragments. Individuals received labels according to their group identity as detected with STRUCTURE, restricted to group membership probabilities exceeding 0.7. Colour codes are explained in the accompanying figure title. The axes represent the first two principal coordinates (TIFF 6699 kb)
Online Resource 8
Parent-specific recombination and LD. Illustration of the effect of differential parental recombination on the LD decay of chromosomes 2 to 12 respectively. The left graph depicts the LD decay for the paternal and maternal markers together, the middle plot shows the decay for paternal markers exclusively (RH) and the right diagram presents the decay for the maternal markers only (SH). The title of each plot indicates the exact number of markers on which the decay is based (DOC 2,275 kb)
Online Resource 9
Group-specific LD patterns for chromosomes 2 to 12 respectively. From left to right LD decay plots are presented for four groups previously discovered with STRUCTURE and restricted to cultivars with group membership probabilities exceeding 0.7. For each plot, the title names first the group itself (Anc = Ancient, Fre = Fresh consumption, Pro = Processing and Sta = Starch) followed by the exact number of cultivars allocated to the group. In each case markers from both paternal and maternal map were combined, the total number of markers is mentioned in the title. Horizontal lines in each plot represent the calculated significance thresholds for the 0.95 quantile (striped) and the 0.99 quantile (full) (DOC 3,759 kb)
Online Resource 10
Heat map visualising the LD pattern along the potato genome based on genotypic data of 53 microsatellites. R2 was used as LD measure to construct the map. Labels consist of the name of the microsatellite followed by its chromosome number. Markers are ordered according to their relative position on the respective chromosomes. A blue-to-red colour spectrum indicates the intensity of LD between pair-wise combinations of SSR loci as explained in the figure legend (TIFF 515 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
D’hoop, B.B., Paulo, M.J., Kowitwanich, K. et al. Population structure and linkage disequilibrium unravelled in tetraploid potato. Theor Appl Genet 121, 1151–1170 (2010). https://doi.org/10.1007/s00122-010-1379-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1379-5