
Abstract
Ultra-performance liquid chromatography combined with time-of-flight mass spectrometry (UPLC–ToF-MS) has been used for screening and quantification of more than 100 veterinary drugs in milk. The veterinary drugs represent different classes including benzimidazoles, macrolides, penicillins, quinolones, sulphonamides, pyrimidines, tetracylines, nitroimidazoles, tranquillizers, ionophores, amphenicols and non-steroidal anti-inflammatory agents (NSAIDs). After protein precipitation, centrifugation and solid-phase extraction (SPE), the extracts were analysed by UPLC–ToF-MS. From the acquired full scan data the drug-specific ions were extracted for construction of the chromatograms and evaluation of the results. The analytical method was validated according to the EU guidelines (2002/657/EC) for a quantitative screening method. At the concentration level of interest (MRL level) the results for repeatability (%RSD < 20% for 86% of the compounds), reproducibility (%RSD < 40% for 96% of the compounds) and the accuracy (80–120% for 88% of the compounds) were satisfactory. Evaluation of the CCβ values and the linearity results demonstrates that the developed method shows adequate sensitivity and linearity to provide quantitative results. Furthermore, the method is accurate enough to differentiate between suspected and negative samples or drug concentrations below or above the MRL. A set of 100 samples of raw milk were screened for residues. No suspected (positive) results were obtained except for the included blind reference sample containing sulphamethazine (88 μg/l) that tested positive for this compound. UPLC–ToF-MS combines high resolution for both LC and MS with high mass accuracy which is very powerful for the multi-compound analysis of veterinary drugs. The technique seems to be powerful enough for the analysis of not only veterinary drugs but also organic contaminants like pesticides, mycotoxins and plant toxins in one single method.
Similar content being viewed by others


Introduction
Incorrect use of drugs in veterinary practice may leave residues in edible tissues. These residues may have direct toxic effects on consumers e.g. allergic reaction in hypersensitive individuals, or for example antibiotics may cause problems indirectly through induction of resistant strains of bacteria. The EU has set maximum residue limits (MRLs) for a variety of veterinary drugs in tissues, milk and eggs [1]. In order to detect such residues in food and tissues, microbiological or bioassay techniques are widely used as screening methods. These methods generally do not distinguish between members of a class of antibiotics, but provide a semiquantitative estimate of ‘total’ residues detected. Nevertheless, they continue to be used because of their simplicity and low cost.
The cost-effectiveness of analytical procedures is becoming an important issue for all laboratories involved in residue analysis. A way to improve cost-effectiveness is to maximise the number of analytes that may be determined by a single procedure or from a single portion of test material. Such an approach is extremely effective when multi-compound techniques, such as liquid chromatography in combination with triple-quadrupole mass-spectrometric detection (LC–QqQ-MS) are used. However, most reported multi-compound methods target a few closely related compounds, usually belonging to a single drug class [2, 3]. There are a few procedures describing methods that can analyse compounds from unrelated classes of drugs [4–6]. However, increasing the number of analyte peaks to be monitored beyond 100–120, requires multiple injections or monitoring specific transitions at a specific retention time window [7]. Methods based on retention time windows, require frequent readjustments due to small shifts of retention times. An attractive alternative is the use of a full mass scan MS technique, for example by using time-of-flight (ToF) MS. The medium to high resolution of ToF-MS of 10,000 FWHM effects a significant selectivity and therefore sensitivity gain compared with unit-resolution scanning MS instrumentation. An advantage of ToF-MS is that no a priori hypothesis about the presence of certain drugs is required; that is, no analyte-specific transitions have to be defined, as is necessary before injecting a sample onto an LC–QqQ-MS using multiple reaction monitoring (MRM). The high-resolution, full scan data permit the testing of any a posteriori hypotheses by extracting any desired exact mass chromatogram. Moreover, the accurate mass capability of LC–ToF-MS allows the reconstruction of highly selective accurate mass chromatograms of target residues in complex matrices. Accurate mass determination and calculated elemental composition data can be used for structure elucidation as well [8].
In the multi-compound method using the full scan MS, the LC part of the system sometimes causes certain limitations. Due to the high number of compounds to be separated the run times will become relatively long. Novel low-dead-volume, high-pressure (1,000 bar) LC equipment provides strategies to improve resolution while maintaining or even shortening run times. This technique is called ultra-performance LC (UPLC). An essential aspect of the UPLC concept is the use of sub-2-μm particulate packing materials, while maintaining other aspects of the column geometry, e.g. column length. UPLC–ToF-MS provides significant advantages concerning selectivity, sensitivity and speed. The higher resolution provided by UPLC is an important factor to compensate for the fact that the selectivity of currently available ToF-MS instrumentation is still less than that provided by monitoring MS→MS transitions.
A very interesting example of the use of UPLC combined with full scan mass MS for multi-compound screening of veterinary drugs in urine is described by Kaufmann et al. [9]. The method covers more than 100 analytes belonging to different classes of veterinary drugs. The quantification limits were < 10 μg/l for over 90% of the 108 tested veterinary drugs and metabolites.
This paper describes the validation of a comprehensive screening and quantification method based on UPLC–ToF-MS for the analysis of 101 veterinary drugs in milk. The selected veterinary drugs represent 12 classes of drugs. The benzimidazoles belong to the group of anthelmintic drugs acting primarily against lungworms and liver fluke. The penicillins, macrolides, quinolones, sulphonamides, tetracyclines and amphenicols are different classes of antibiotics. The nitroimidazoles and ionophores are frequently used as feed additives for the treatment and prevention of certain bacterial and protozoal diseases. Finally the tranquillizers and the non-steroidal anti-inflammatory agents (NSAIDs) are included in the method. Milk was selected because it is an important matrix for residue control which can contain residues from different classes of veterinary drugs. UPLC–ToF-MS was used to screen a series of milk samples for drug residues. Both qualitative and quantitative results obtained during validation and the results obtained by the analysis of 100 different samples of raw milk will be presented and discussed.
Materials and methods
Chemicals, reagents and solutions
Veterinary drug analytical standards were purchased from Sigma-Aldrich Chemie b.v (Zwijndrecht, the Netherlands) SmithKline Beecham (Zeist, the Netherlands), Riedel-de Haen (Seelze, Germany) or Fluka (Buchs, Switzerland). Oxfendazole and oxfendazole sulfone were purchased from Syntex (Clare Castle, Co Clare, Ireland), valnemulin from Novartis (Basel, Switerzerland) and marbofloxacin from Vetoquinol (New Jersey, VS). Albendazole (sulfoxide), hydroxyipronidazole, carazolol, propyphenazone and piroxycam were obtained from Bundesamt fur Verbraucherschutz und Lebensmittelsicherheit, BVL-CRL (Berlin, Germany). 5-Hydroxythiabendazole was obtained from The National Institute for Public Health and the Environment, RIVM-CRL (Bilthoven, the Netherlands). Figure 1 presents the structures of a subset of the veterinary drugs studied.

Structures of representative compounds from each class of drug studied
Individual veterinary drug stock solutions (0.1, 1 or 2 mg/ml; azaperone 10 μg/ml) were prepared in pure methanol or acetonitrile and were stored at −18 °C. The mixed standard solution was stored at −18 °C and was used for 1 week. Sub-portions of the mixed standard solution are stored at −80 °C; every week a portion of this mixed standard solution was taken and used for analysis. The penicillins are not very stable drugs and these compounds were therefore added to the mixed standard solution just before analysis. The selected compounds, see Table 1, comprise the major classes of veterinary drugs that are commonly used to treat diseases in veterinary practice. Cinchophen (C16H11NO2; retention time (Rt) = 3.99 min) and nigericine (C40H67O11; Rt = 8.44 min) sodium salt were used as the internal standards. The internal standards were not used for recovery corrections but for monitoring the efficiency of the extraction procedure and to monitor the run-to-run differences in retention times. LC/MS-grade acetonitrile, water and methanol were obtained from Biosolve (Valkenswaard, the Netherlands). Formic acid was obtained from Merck. Solid-phase extraction columns (Strata-X 33-μm polymeric reversed-phase 60 mg/3 ml) were obtained from Phenomenex (Torrance, CA, USA).
Samples
All milk samples tested were raw (not pasteurised) milk samples collected by the Dutch Food and Consumer Product Safety Authority, Laboratory Region East (Wageningen, The Netherlands), at farmhouses during the spring of 2007. The samples were received frozen and were kept frozen (−20 °C) until analysis.
The reference sample of milk used was previously used in a proficiency test organized by Progetto Trieste (Laboratory Proficiency Testing for Food Analysis; Italy). The sample was coded Progetto/2007/Mi329A. The assigned value was 87.8 μg/l sulphamethazine (=sulphadimidine).
Instrumentation
Liquid chromatography
The separation of the veterinary drugs from the raw milk extracts was carried out using an ultra-performance liquid chromatography (UPLC) system, consisting of a vacuum degasser, autosampler and a binary pump (Acquity UPLC system; Waters, Milford, MA) equipped with a reversed-phase Waters acquity UPLC BEH C18 analytical column of 50 × 2.1 mm and 1.7-μm particle size. The gradient (solvent A, water/formic acid (1,000:1, v/v); solvent B, water/acetonitrile/formic acid (100:900:1, v/v/v)) was 0 min, 0% B; 1–6 min, linear increase to 40% B; 6–7.5 min linear increase to 100% B with a final hold for 1 min. Injection volume was 40 μl. The flow rate was 800 μl/min and was split (1:7) before entering the ToF.
LC–electrospray time-of-flight mass spectrometry
The UPLC system was connected to a time-of-flight mass spectrometer Waters-Micromass LCT Premier ToF equipped with an electrospray interface operating in the positive ion mode, using the following parameters: cone voltage, 50 V; capillary voltage, 2,800 V. Full scan spectra from 100 to 1,000 Da were acquired with a scantime of 0.2 s. Mass accuracy was maintained by using a lock spray with lock mass of leucine-enkefaline 12C [M+H]+ ion m/z 556.2771. Resolution was at least 10,000 FWHM at m/z of the lock mass. Dynamic range enhancement (DRE) was switched on.
Sample pre-treatment and extraction
The milk (2 ml) was mixed with acetonitrile (2 ml) to effect precipitation of proteins. After an intensive shaking period of 30 min, the samples were centrifuged for 15 min (3,600 g at T = 10 °C). From the supernatant 2 ml was diluted ten times with water. The diluted sample was applied to a StrataX-SPE column. The column was conditioned with 3 ml methanol and 3 ml water and washed with 3 ml water. The analytes were eluted with 3 ml methanol.
UPLC–ToF-MS analysis
After evaporation of the StrataX eluate under a stream of nitrogen at 40 °C, the dried extract was redissolved in 50 μl acetonitrile and vortexed for 30 s. Next 450 μl water/formic acid (1,000:2, v/v) was added and an aliquot of 40 μl was analysed by UPLC–ToF-MS in the full scan mode. After acquisition the specific [M+H]+ ions (see Table 1) were extracted from the spectra. The used extraction mass window width was drug specific and ranged from 10 to 200 mDa.
Quantification
For quantification, a detector response (peak areas) versus concentration plot was constructed. To this end, five blank milk samples were fortified with different concentrations of the specific drug. The real milk samples were analysed together with the calibration samples (matrix-matched standards or MMS); concentrations were calculated using the linear regression method.
Validation
The developed method was validated based on the procedure described in ref. [10] for quantitative screening. The following characteristics have to be determined: repeatability, within-lab reproducibility, accuracy, linearity, CCβ, selectivity/specificity, robustness and stability. CCβ is defined as the smallest content of the substance that may be detected, identified and/or quantified in a sample with an error probability of β (in this study β = 5%).
Repeatability, within-lab reproducibility, accuracy
The validation study for the quantitative screening of veterinary drugs in milk was carried out at three concentration levels viz. 0.5–1.0–1.5 times the validation level (VL), defined in this paper as the MRL or recommended concentration level recommended by EU Community Reference Laboratories [10]. For drugs without an MRL or recommended concentration level a specific level of interest was defined based on the drug characteristics (class of compounds) or based on MRL or recommended concentration level of the specific drug in other matrices like liver, kidney or meat. All concentration levels used for the validation study are described in Table 1. For some compounds the VL was at a slightly higher level than the MRL or recommended level due to the limited sensitivity of the ToF detector for that specific drug. Some compounds are validated at a concentration level below the MRL or recommended level to prevent the amount of compound detected being outside the range for which the ToF is linear. Blank samples of milk were fortified with a specific concentration of drug and seven replicates of each sample were analysed on 1 day. The procedure was repeated on two additional days.
From the data obtained intra-day repeatability, within-lab (inter-day) reproducibility and accuracy were calculated. Preferably the minimum %RSD for the repeatability and the within-lab reproducibility have to be as described in Table 2 [11]. The accuracy, i.e. (detected concentration divided by added concentration) × 100%, preferably has to be within the 70–120% range. This range is set for this study as an acceptable accuracy for a multi-compound quantitative screening method for the concentration range 1–150 μg/l.
Linearity
The linearity was determined for a concentration range of 0–0.25–0.5–1–2 and 4 times the VL. On each validation day the calibration curves were constructed by means of plotting the detection response of the matrix-matched standard solutions versus the concentrations by means of regression analysis. From these data the regression coefficients (R 2) of the calibration curves were calculated. The criterion for good linearity was R 2 > 0.99.
CCβ and LOQ
The detection capability (CCβ) at the VL was determined as follows: the CCβ = CCα+1.64 × sd(at VL) (sd = standard deviation of within-lab reproducibility) in which CCα = VL+1.64 × sd(at VL).
Additionally to the CCβ the limit of quantification (LOQ) was determined by analysing seven samples of milk fortified at the concentration level of 1/8 × VL. In cases where the signal/noise at this concentration was ≥6, 1/8 × VL was set as the LOQ; otherwise the next calibration level with signal/noise ≥ 6 was set as the LOQ.
Robustness
The robustness of the method was tested by analysing four samples of milk in duplicate and for each sample the sample pre-treatment/extraction procedure was slightly different. The first sample was analysed by using the developed procedure. For the second sample the extraction time was extended from 30 to 60 min. For the third sample the centrifugation step was at 3,600 rpm instead of 3,600 g. For the fourth sample the final residue after evaporation of the solvent was kept dry for 30 min before resolution. The method is robust in the case the %RSD of a specific compound within these eight analyses ≤ %RSD of the corresponding within-lab reproducibility.
Specificity
The specificity of the method was checked by the analysis of 20 blank samples. The chromatograms were monitored for peaks interfering with those of the drugs of interest.
Stability
No stability experiments were performed in this study. The stability data were obtained from previous studies carried out for the specific classes of drugs during the validation study of the LC–QqQ-MS methods. From these studies it is known that all compounds of interest were stable for at least 1 month with the exception of the penicillins. Penicillins are not very stable and their standard solutions have to be prepared right before the validation experiments.
Sample analysis
The samples of raw milk were analysed in four series of 25 samples each. Each series of samples started and ended with the analysis of the matrix-matched calibration standards. The samples were analysed by the method and the experimental conditions as described above.
The criteria for acceptance of the analytical results were:
-
Sensitivity check (signal noise at LOQ level ≥ 6)
-
Deviation of r of the two calibration curves ≤ 20%
-
Relative retention time of suspected analyte and reference standard within the tolerance interval of ±2.5%
-
Criterion for good linearity: R 2 > 0.99
In the case of a suspected result (positive response) the sample was reanalysed for the specific compound(s). A suspected result is obtained when the response measured for an MRL substance is at or above the CCβ level and for all other substances at or above the LOQ level. The confirmatory analysis is based on an LC–QqQ-MS method monitoring two product ions of the suspected compound(s) thereby fulfilling the criteria described by the guidelines [11] for confirmatory analysis.
Results
General considerations
Sample pre-treatment
Different sample pre-treatment/extraction approaches for the determination of residues of drugs in milk are described in the literature and are in use in our laboratory. Possible approaches are liquid/liquid extraction (LLE), solid-phase extraction (SPE), combination of LLE and SPE, ultrafiltration, etc. [4]. For the described multi-compound method the sample pre-treatment step has to be very generic. Therefore ultrafiltration and polymer-based C18-SPE columns (Oasis and StrataX) were tested. The use of ultrafiltration did not show satisfactory recoveries. The use of SPE resulted (due to less matrix interferences and a concentration step) in higher accuracies and lower detection limits. It is obvious that by introduction of an SPE-C18 step the very polar compounds will not be recovered; however, on the other hand by using SPE the extract is concentrated which makes it possible to also detect prohibited compounds at very low levels. The recovery results obtained by using the StrataX SPE column were 5–10% higher (depending on the analyte) than those obtained for the Oasis SPE; therefore the StrataX SPE column is used for the multi-screening method.
UPLC–ToF-MS screening method
The UPLC–ToF-MS full scan accurate mass screening procedure enables the analysis of more than 100 veterinary drugs and metabolites. The main advantage of the proposed approach is the theoretically unlimited number of compounds to be screened simultaneously at low concentration levels.
To construct the screening method, a solvent-based standard with the mixture of studied veterinary drugs was analysed. The method is constructed based on the retention times and responses at specific accurate masses. In the method the different combinations of retention time and accurate mass are defined with their respective acceptable tolerances. After analysis of a real sample the full scan chromatogram is processed and thereby a list of detected compounds (included in the screening method) is generated. A big advantage of the UPLC–ToF-MS method is that not all compounds of interest a priori have to be defined. If later in time a specific compound becomes of interest (which was not included in the screening method) the collected full scan spectra is reprocessed and checked for presence of that specific new compound.
QqQ MS confirmation method
For each class of drug, analytical methods are available for the quantification and identification based on LC–QqQ-MS [4, 12]. By the use of the UPLC–ToF-MS technique a real multi-compound method including different classes of drugs becomes available. For the confirmation of identity of veterinary drugs in products of animal origin the criteria as described in ref. [11] for confirmation have to be met. The confirmation is based on the collection of so-called identification points (IPs). For the confirmation of the identity of unauthorised substances a minimum of four IPs is required. For the confirmation of the identity of MRL substances a minimum of three IPs is required. The number of IPs earned by a specific analysis depends on the technique used. However, almost invariably these techniques have to be based on mass spectrometric detection. A low-resolution mass spectrometer, such as a QqQ or an ion-trap (IT), is able to acquire 1.0 IP for the precursor ion and 1.5 IPs for each product ion; that is, with the selection of two multi-reaction-monitoring (MRM) transitions, 4.0 IPs are acquired. The mass accuracy of a high-resolution mass spectrometer (resolution ≥ 20,000 FWHM) acquires 2.0 IPs for the precursor ion and 2.5 for each product ion, so, in this case, the IPs acquired in the product ion scan mode are (2+2.5n). However identification criteria for the analysis by ToF-MS, medium to high resolution of approximately 10,000 FWHM, are not included (yet) in the EU document [11]. Therefore UPLC–ToF-MS is used as a quantitative screening method. The samples suspected of containing a drug at a concentration level above the MRL or MRPL will be analysed by an LC–QqQ-MS method for the final confirmation of the identity.
Accurate mass measurements
Table 1 presents the accurate mass measurement data obtained for milk extracts. In general for a ToF having a mass resolution of ca. 10,000 FWHM and an external calibration, a deviation of the measured accurate mass versus the calculated mass of 10 ppm is acceptable [13], especially when the low concentration levels (1–150 μg/l) are taken into account. From Table 1 we concluded that the accurate measurements in a real sample extract are acceptable for more than 80% of the compounds studied. It is noted that 15 out of 21 compounds with a measured mass deviation > 10 ppm elute with a retention time > 5.5 min. Looking at the total ion current (TIC) chromatogram of a milk extract (Fig. 3) it is obvious that beyond 5.5 min most of the matrix compounds are eluting. For the analysis an absolute amount of 1 ml of milk is concentrated to a final volume of 500 μl from which 40 μl is injected, corresponding to approximately 80 mg of matrix equivalent per injection. For the UPLC–ToF-MS method described in ref. [9] only 0.4 μg of matrix was injected. The unexpected low mass accuracy for compounds eluting above 5.5 min is probably due to elution of a large amount of matrix compounds [14]. However, the concentration step is necessary to detect the unauthorised compounds at the 1 μg/l level.
Validation study
The results obtained at the different concentration levels (0.5–1–1.5) × VL are in the same range. Therefore only the results obtained for 1 × VL are presented in Table 1. The exceptions are discussed below.
Repeatability, reproducibility and accuracy
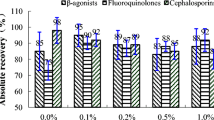
We concluded that at the validation level (VL) more than 65% of the compounds tested show a repeatability of RSD < 10% and less than 15% of the compounds show a repeatability of RSD > 20%. At 0.5 × VL the percentages are significantly different. At 0.5 × VL almost 50% of the compounds show repeatability results of RSD > 10%. Comparing the 1 × VL and 1.5 × VL levels only slightly better results are obtained for the latter. At the 1 × VL level 34 compounds show RSD > 10% and only 13 compounds show RSD > 20%. Three compounds out of the group of tranquillizers show RSD > 20%, probably due to the low concentration of interest (< 5 μg/l). Furthermore the more polar and early-eluting compounds like isopyrin and sulphisoxazole show RSDs > 20%. Overall the repeatability results are acceptable (RSD < 20%) for more than 86% of the compounds.
From the within-lab reproducibility results we concluded that 19 compounds show reproducibility results RSD > 20% at the 1 × VL. There are four compounds with unacceptably high (> 40% at 1 × VL) reproducibility results. These results are obtained for two tranquillizers, propionylpromazine and chloropromazine, for the non-steroidal anti-inflammatory drug (NSAID) phenylbutazone and for the ionophore salinomycine. We concluded that for 96% of the compounds the reproducibility results are satisfactory (%RSD < 40%).
From the accuracy results we concluded that 89% of the compounds show acceptable accuracy results. There are only a few compounds with accuracy results < 80% or > 120% even at the 1 × VL. Compounds with an accuracy > 120% are levamisole, (hydroxy)thiabendazole, lincomycine, ampicillin, ipronidazole, phenylbutazone and florphenicol. The compounds norfloxacin and danofloxacin show accuracies of 121%. The compound with an accuracy (at 1 × VL) < 70% is not surprisingly the unstable nafcillin. The accuracy of chloropromazine is not established. For this compound no accurate quantification is possible at the concentration range of interest.
For compounds with a repeatability of RSD > 20%, reproducibility RSD > 40% and/or accuracy of >1 20% or < 70%, accurate quantification is not possible. For these compounds accurate quantification is only possible by applying a standard addition procedure or after the method has been optimized for the specific compound.
CCβ, LOQ and linearity
After evaluation of the results some general conclusions can be drawn. The first is that for the compounds with 1 × VL between 1 and 50 μg/l the CCβ are all within the range of 1.2–100 μg/l (see Table 1). Three drugs, lincomycine, sulphisoxazole and isopyrin, show CCβ values of > 100 μg/l at 1 × VL of 50 μg/L. For the tetracyclines the 1 × VL values are 100 μg/l and the CCβ range from 129.9–141.8 μg/l.
For chloropromazine no CCβ is established for the above-described reason.
The second conclusion is that the LOQ levels (not presented here) for 93 of the 101 compounds are at or below 1/8 × VL. For azaperol and naproxen the LOQs are 1/4 × VL and for isopyrin and three of the ionophores the LOQs are 1/2 × VL. For the ionophore salinomycine and for phenylbutazone the LOQ is at 1 × VL. For some drugs the 1 × VL was set a little higher than the MRL or recommended concentration. Taking this into account only three NSAIDs, diclofenac, naproxen and phenylbutazone, cannot be detected at the recommended concentration. The LOQ of diclofenac is only slightly higher, namely 6.3 μg/l (recommended 5 μg/l). The LOQ of naproxen is 12.5 μg/l (recommended 10 μg/l) and for phenylbutazone 25 μg/l (recommended 5 μg/l). When the LOQs are compared with the results presented by Kaufmann et al. [9] for urine samples (> 90% of the compounds LOQ < 10 μg/l) we concluded that in this respect the presented method for milk is even more sensitive because > 90% of the compounds tested show LOQs of < 7 μg/l. It has to be mentioned that for the presented method a SPE step was included. Kaufmann only used a dilution step.
A third conclusion is that the regression coefficients (R 2) (not presented here) of approximately 80% of all matrix-matched calibration curves versus concentration plots ranged from 0.25 × VL to 4 × VL are > 0.99. Moreover, all curves show R 2 > 0.9 with only three exceptions, lincomycine with a R 2 of 0.87, isopyrin with R 2 of 0.86 and again chloropromazine for which no R 2 is established.
Robustness, specificity/selectivity
From the %RSD within the robustness samples we concluded that the method was robust (%RSD robustness ≤ %RSD within-lab reproducibility). The method is also specific/selective, since no peaks (>LOQ) were detected in known blank samples of milk.
Application study
The 100 milk samples were analysed on four different days. For all days the sensitivity and linearity checks were OK. None of the samples was marked as suspected of containing a veterinary compound. However the included reference sample tested positive for sulphamethazine (Fig. 2). The technician did not know that this reference sample was included (blind sample). The reference sample was previously analysed by an LC-QqQ-MS method for containing sulphonamides and tested positive for sulphamethazine (86 μg/l assigned value 88 μg/l). With the current screening method the concentration was approximately 82 μg/l.

UPLC–ToF-(ESI+)MS chromatogram of proficiency test sample of milk with a selection of extracted ion chromatograms; sample contains sulphametazine (88 μg/l); extraction mass window 0.020 Da
All the samples (except the reference sample) were previously screened negative for the antibiotics (tetracylines, aminoglycosides, macrolides and penicillins) by using microbiological screening tests. Microbiological screening tests are group specific. The UPLC–ToF-MS method is compound specific (individual drugs are identified). Furthermore it is very easily to add a new drug even from another class of drugs to the UPLC–ToF-MS method and retrospective analysis for a specific compound is possible by simply reprocessing the acquired data construction of the extracted ion chromatogram (XIC).
Hardly any differences can be observed between the total ion chromatogram (TIC) obtained for a blank and the fortified sample. However, looking at an example of a typical XIC of the blank presented in Fig. 3a and the fortified sample of milk (at 1 × VL) presented in Fig. 3b the differences are clear. The extracted ions are a representative sub-selection of the compounds tested, including: albendazole m/z 266.0963; thiabendazole m/z 202.0439; flumequine m/z 262.0879; norfloxacin m/z 320.1410; enrofloxacin m/z 360.1723; sulphadiazine m/z 251.0602; sulphamethoxazole m/z 254.0599; tetracycline m/z 445.1611; diclofenac m/z 296.0250.


Typical example of a UPLC–ToF-(ESI+)MS extracted ion chromatograms of a blank sample of milk and b blank sample fortified at the 1 × VL; extraction mass window 0.050 Da, and c = b with extraction mass window of 0.010 Da (see text for more details)
Discussion
From the literature it is known that the selected width of the extraction mass window is important. When a very small extraction mass window width is selected there is possible chance of false negative results [15]. Co-elution of isobaric compounds can result in significant deviations in exact mass measurements. The accurate mass assigned to a peak is the average of the accurate masses of the co-eluting peaks. Therefore it is possible that the average accurate mass is outside the extraction mass window width which is set for the specific compound of interest. The final result is a false negative observation [15]. When the extraction mass window is set at 0.05 Da (Fig. 3a,b) all peaks are detected. When the extraction mass window is set at 0.01 Da as is shown in Fig. 3c, all the peaks are still detected showing exactly the same abundances with thiabendazole (m/z 202.4399) being the only exception. The thiabendazole peak splits into two separate peaks at this low extraction mass window width. This observation is possibly not due to an isobaric interfering compound co-eluting with thiabendazole because in the blank sample no peak is detected. It is expected based on the shape of the peak in Fig. 3b, that this peak shows high intensity and that the detector is overloaded. When this occurs the dynamic range enhancement (DRE), which is a technical device within the ToF, will electronically reduce the signal. This functionality does not influence the acquired data; however, sometimes this ‘correction’ of signal results in a ‘dip’ at the top of the peak. In summary the peak is split probably due to the effect of the DRE.
The influence of the extraction mass window width is more obviously demonstrated in Fig. 4. Figure 4 presents the full scan TIC chromatogram of a sample of milk. To find out if this sample contains oxolinic acid the XIC of the ion m/z 262.0715 is constructed. Two peaks are detected. Reducing the width of the extraction mass window from 1 to 0.050 Da still results in two peaks being detected, one at Rt = 3.96 min and one at Rt = 4.95 min. Only when the extraction mass window is set at 0.010 Da is one peak, oxolinic acid, at Rt = 3.96 min, detected. The peak detected at Rt = 4.95 is flumequine with an accurate mass of m/z 262.0879. In this typical example, also described by Kaufman for urine samples [9], the drugs can easily be separated based on the retention times; however when this is not the case, for example when less selective LC is used, the two compounds can only be separated from each other when the mass resolution of the ToF is high enough. The ion mass differs by 262.0879–262.0715 = 0.0164 Da. The mass resolutions of the detector in combination with mass accuracy are important parameters for a reliable screening result [14, 16].

Typical UPLC-ToF-(ESI+)MS chromatogram for a milk sample fortified with 2 μg/l oxolinic acid and 50 μg/l flumequine; effect of different extraction mass windows (see text for details)
Some authors sometimes confuse the term extraction mass window width and mass resolution [9]. These are two completely different things. The mass resolution is the power of the ToF-MS to separate two ions with only slightly different accurate masses. The extraction mass window width has nothing to do with MS resolution; it is just a parameter used to present the detected ions, in other words it defines the mass range of the ions which has to be extracted from the acquired data to construct the chromatogram.
From the examples given above it is concluded that UPLC–ToF-MS is a very powerful technique for multi-compound detection and in theory for an unlimited number of compounds [17]. Therefore in our lab research is ongoing to use the described method not only for the determination of veterinary drug residues but also to include other organic residues and contaminants like pesticides, environmental contaminants, mycotoxins and plant toxins. The first experiments have been performed and the first preliminary results obtained. The samples of milk are extracted with an acetonitrile/formic acid mixture; after a centrifugation step the extracts are directly injected into the UPLC–ToF-MS and screened for more than 270 compounds. The results of this comprehensive analysis of residues and contaminants are very promising and will be published separately elsewhere [18].
Conclusions
There is increasing interest in methods for the simultaneous analysis of various classes of veterinary drugs. Such multi-compound analyses which, in one run, can deal with more than 100 compounds are only possible by using full mass scan MS techniques like ToF-MS. Several examples have already been published, with UPLC–ToF-MS probably being the most powerful measurement tool as is also demonstrated by the present study. The use of UPLC is especially powerful when biological extracts have to be screened because the additional LC selectivity compensates for the lack of selectivity in comparison with the MS/MS option of a QqQ-MS.
In the past, an LC–QqQ method (utilizing selected MRM transitions) was the starting point for method development. Today, in theory all compounds can be measured by full scan MS. The starting point now is no longer the detection conditions used, but the sample material. Starting with for example milk all relevant veterinary drugs—irrespective of the class they belong to—can be detected in one (UP)LC–ToF-MS analysis, and the most important part of the method development is the generic extraction of the compounds of interest out of the matrix. This will no doubt cause an increased interest in a variety of modern sample pre-treatment techniques. The sample pre-treatment step becomes even more interesting when other organic contaminants like residues of pesticides, mycotoxins and or plant toxins are included in the comprehensive screening method.
In summary, the UPLC–ToF-MS combination offers unsurpassed performance for screening purposes, it can also effectively provide concentration values, and is accurate enough to differentiate between positive and negative samples or drug concentrations below or above the MRL.
Finally it has to be mentioned that the confirmatory analysis of the suspected samples has to be done by the use of an MS/MS technique because criteria for confirmation of the identity of drugs by ToF-MS are not included yet in the EU guidelines.
The introduction of new MS techniques like ToF also has consequences for the EU criteria defined for confirmation of compound identity. Both mass resolution and mass accuracy data are of influence for such identity confirmation and both parameters should preferably be included in the revised version of Commission Decision 2002/657/EC.
References
European Union (1990) Council Regulation 2377/90/EC of 26 June 1990 laying down a Community procedure for the establishment of maximum residue limits of veterinary medicinal products in foodstuffs of animal origin. OJEU L224 18 August 1990
Danaher M, De Ruyck H, Crooks SRH, Dowling G, O’Keeffe M (2007) J Chromatogr B 845:1–37
Stolker AAM, Brinkman UATh (2005) J Chromatogr A 1067:15–53
Granelli K, Branzell C (2007) Anal Chim Acta 586:289–295
Yamada R, Kozono M, Ohmori T, Morimatsu F, Kitayama M (2006) Biosci Biotechnol Biochem 70:54–65
Stolker AAM, Zuidema T, Nielen MWF (2007) Trend Anal Chem 26:967–979
Muñoz P, Blanca J, Ramos M, Bartolomé M, García E, Méndez N, Gomez J, de Pozuelo MM (2005) Anal Chim Acta 529:137–144
Nielen MWF, van Engelen MC, Zuiderent R, Ramaker R (2007) Anal Chim Acta 586:122–129
Kaufmann A, Butcher P, Maden K, Widmer M (2007) Anal Chim Acta 586:13–21
CRL Guidance Paper (2007) CRLs view on state of the art analytical methods for national residue control plans. Community Reference Laboratories (CRLs) for residues according to Council Directive 96/23/EC
European Union (2002) Commission Decision (2002/657/EC) of 12 August 2002. OJEC L221:8–36
Turnipseed SB, Andersen WC, Karbiwnyk CM, Madson MR, Miller KE (2008) Rapid Commun Mass Spectrom 22:1467–1480
Ojanperä S, Pelander A, Pelzing M, Krebs I, Vuori E, Ojanperä I (2006) Rapid Commun Mass Spectrom 20:1161–1167
Calbiani F, Careri M, Elviri L, Mangia A, Zagnoni I (2006) J Mass Spectrom 41:289–294
Kaufmann A, Butcher P (2006) Rapid Commun Mass Spectrom 20:3566–2572
Laures AMF, Wolff JC, Eckers Ch, Borman PhJ, Chatfield MJ (2007) Rapid Commun Mass Spectrom 21:529–535
Garcia-Reyes JF, Hernando MD, Ferrer C, Molina-Diaz A, Fernandez-Alba AR (2007) Anal Chem 79:7308–7323
Mol HJM et al (2008) Anal Bioanal Chem (in press)
Acknnowledgement
This study was financial supported by the Dutch Ministry of Agriculture, Nature and Food Quality (project # 77163901).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Stolker, A.A.M., Rutgers, P., Oosterink, E. et al. Comprehensive screening and quantification of veterinary drugs in milk using UPLC–ToF-MS. Anal Bioanal Chem 391, 2309–2322 (2008). https://doi.org/10.1007/s00216-008-2168-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-008-2168-8