
Abstract
The anonymous marker systems microsatellites (simple sequence repeats), amplified fragment length polymorphisms and sequence-specific amplified polymorphisms were compared with the targeted marker systems sequence-related amplified polymorphisms, target region amplification polymorphisms and nucleotide binding site profiling for their ability to describe the genetic diversity in a selected set of 80 Lactuca accessions. The accessions were also described morphologically, and all characterisation methods were evaluated against the genetic diversity assessed by a panel of three crop experts. The morphological data showed a low level of association with the molecular data, and did not display a consistently better relationship with the experts’ assessments in comparison with the molecular data. In general, the diversity described by the targeted molecular markers did not differ markedly from that of the anonymous markers, resulting in only slight differences in performance when related to the expert-based assessments. It was argued that markers targeted to specific gene sequences may still behave as anonymous markers and that the type of marker system used is irrelevant when at low taxonomic levels a clear genetic structure is absent due to intensive breeding activities.
Similar content being viewed by others


Introduction
Over the last decennia, many genebanks have been established to conserve genetic diversity for present and future utilisation. It is generally agreed that the main goal of genebanks is to constitute collections of genetic resources that represent as wide as possible the genetic diversity of a crop gene pool with a minimum of redundancy (Frankel and Brown 1984). However, capturing the entire diversity of a crop gene pool in a collection is obviously unrealistic. Therefore, genebank curators sample the gene pool by trying to select material with as wide as possible morphological and agronomical variations. Because these data are often initially lacking, curators generally use secondary data, such as taxonomic and eco-geographical data, to select material for inclusion in their collections. However, this remains a rather intuitive process based on incomplete and often unreliable data (Engels and Visser 2003). Therefore, characterisation data are usually collected a posteriori in order to obtain insight into collection composition and subsequently to optimise the genetic diversity thereof.
For a long time, germplasm characterisation was carried out solely by morphological description. Nowadays, molecular marker technologies are increasingly being used to complement traditional methods because of their ability to measure diversity directly at the DNA level (Bretting and Widrlechner 1995; Brown and Kresovich 1996; Karp et al. 1997; Spooner et al. 2005). A wide variety of technologies to measure genetic diversity have emerged during the past decades, including microsatellites, amplified fragment length polymorphism (AFLP) and sequence-specific amplified polymorphism (SSAP). Microsatellites or simple sequence repeats (SSR) represent segments with tandem repeats of 1–6 base pairs, which have a high genomic abundance in eukaryotes. Specific primers are used to amplify microsatellites by the polymerase chain reaction (PCR). Microsatellites are codominant markers that usually display high allelic diversity (Queller et al. 1993). AFLPs are DNA fragments obtained from digestion with restriction enzymes, followed by ligation of oligonucleotide adapters to the digestion products and amplification by PCR. Many restriction fragments are amplified per assay and the resulting electrophoresis profiles are dominantly scored (Vos et al. 1995). SSAPs are DNA fragments amplified by PCR using a primer designed from conserved sequences of a transposable element and a primer based on the presence of a nearby restriction endonuclease site. SSAP can be considered an anchored AFLP approach, resulting in multi-locus electrophoresis profiles that are dominantly scored (Waugh et al. 1997).
Many of the widely used technologies generate anonymous markers because analyses are usually performed on total genomic DNA and the genomic location of the amplified DNA fragments is generally unknown. For most organisms, the largest part of the genome consists of DNA that seems to have no particular biological function, and hence to carry selectively neutral genetic diversity. Therefore, the majority of the marker technologies can be assumed to sample mainly genomic variation that is shaped by random processes, such as genetic drift (Falconer 1981). Based on a meta-analysis of 71 datasets of random markers, molecular measures of genetic diversity revealed only a very limited ability to predict quantitative genetic variability (Reed and Frankham 2001). However, rather than in neutral variation, users of genebank collections are generally interested in variation from expressed (or functional) regions of the genome that may be under selective pressure.
Molecular markers for genes of interest may be identified by genetic mapping studies and by assessment of linkage relationships with specific traits (e.g. Graner et al. 1999). These approaches are common practice in modern plant breeding, as part of marker-assisted selection, but are far too labour-intensive for routine use in germplasm conservation. Moreover, such approaches are usually restricted to one or a few traits, whereas conservationists are interested in the broader genetic diversity. To study variation in subsets of coding regions of the DNA, marker technologies could be applied to messenger RNA (mRNA), instead of total genomic DNA. Extracted mRNA is first transformed to complementary DNA (cDNA) and subsequently used as template for marker analysis, such as cDNA-AFLP (Brugmans et al. 2002). cDNA markers are used in differential display experiments and as tool in the isolation of genes. However, their value for germplasm characterisation seems rather limited because of the high sensitivity to environmental conditions.
In recent years, novel techniques have become available that target molecular markers to coding regions of the genome, and that seem promising in germplasm characterisation. Sequence-related amplified polymorphism (SRAP) amplifies subsets of open reading frames of genes, resulting in multilocus band profiles that are dominantly scored. In Brassica oleracea L., 45% of the gel-isolated and sequenced DNA fragments could be matched to known genes, and the technique successfully amplified SRAP markers in other crops as well, including lettuce (Li and Quiros 2001). Germplasm characterisation of Cucurbita pepo revealed that, compared to AFLPs, SRAP-based information was more concordant with morphological variability and the evolutionary history of morphotypes (Ferriol et al. 2003). In Buffalograss, SRAPs were found to be valuable tools for efficient germplasm management and breeding programmes (Budak et al. 2004). Target region amplification polymorphism (TRAP) targets ESTs (expressed sequence tags) annotated as putative functional genes. The TRAP technique amplifies multiple fragments per assay that are dominantly scored, and has been successfully tested in multiple crops (Hu and Vick 2003). Application of the technique to the characterisation of Lactuca germplasm demonstrated the ability of TRAP markers to discriminate lettuce cultivars and to group the cultivars by horticultural types (Hu et al. 2005). Nucleotide binding site (NBS) profiling targets the largest class of currently identified plant resistance genes. PCR is carried out with a primer anchored to a conserved sequence of the NBS in combination with a primer based on the presence of a nearby endonuclease restriction site. Electrophoresis patterns of NBS profiling resemble those of AFLP analysis, and are also dominantly scored (van der Linden et al. 2004). For lettuce, sequence analysis of NBS fragments showed that nearly 50% of the markers corresponded to known resistance gene analogues (Syed et al. 2006).
Compared to anonymous markers, such as microsatellites, AFLP and SSAP, markers targeted to potentially functional diversity, such as SRAP, TRAP and NBS may yield more accurate estimates of the genetic diversity that curators and users would like to observe in germplasm collections. To compare the ability of these six marker technologies to describe genetic diversity in a germplasm collection, 80 accessions from the lettuce collection of the Centre for Genetic Resources, The Netherlands (CGN), were characterised. The obtained results were compared to the diversity estimated by morphological description and to diversity estimates based on the knowledge of three experts with a long-lasting experience in different aspects of lettuce diversity, namely crop breeding, variety registration and collection management.
Materials and methods
Study material
The lettuce collection of CGN currently (December 2008) consists of 2,571 accessions, including 1,540 accessions of cultivated material (Lactuca sativa) and 1,031 accessions of crop wild relatives. In order to maximise the genetic diversity within the study, the core selector (van Hintum 1999) was used to select ten accessions from each of the main lettuce crop types butterhead, cos, crisphead, cutting, Latin, oilseed and stalk lettuce (Křístková et al. 2008; Mou 2008), and from prickly lettuce (L. serriola), representing the most important wild relative of cultivated lettuce (Table 1). More detailed accession information can be found at http://www.cgn.wur.nl.
Molecular analyses
Seeds were sown in March 2006 in a greenhouse and after approximately 1 month, about 100 mg of leaf tissue was sampled from a single plant per accession. Leaves were sampled in 2-ml Eppendorf tubes, immediately frozen in liquid nitrogen, and stored at −80°C. Samples were freeze-dried overnight and ground mechanically into a fine powder using a Retch shaking mill. Total genomic DNA was extracted using a combination of the methods described by Fulton et al. (1995) and the DNeasy 96 Plant Kit (Qiagen, Westburg, The Netherlands).
Microsatellites LsA001, LsA004, LsB101, Ls B104, LsD103, LsD106, LsD108, LsD109, LsE003, LsE011 and LsE018 (van de Wiel et al. 1999) were amplified in four multiplex PCR reactions following the methods described by van Treuren et al. (2008). Analyses were carried out on an ABI Prism 3700 DNA Analyzer (Applied Biosystems, Foster City, Calif.). Fragment sizes and peak areas were determined automatically using the GENESCAN analysis software (release 1.1 3700 software, Applied Biosystems), and further processed with the software package Genotyper, version 3.5 NT (Perkin Elmer).
AFLP analysis basically followed the protocol described by Vos et al. (1995). Selective amplification was carried out using the EcoRI primer E35 (selective nucleotides ACA) in combination with each of the MseI primers M49 (CAG) and M59 (CTA) (Jeuken et al. 2001). PCR products radiolabelled with P33 were separated by polyacrylamide gel-electrophoresis. Autoradiograms were manually scored for the presence or absence of AFLP fragments in the range of 50–500 base pairs.
SSAP-analyses followed the methods described by Syed et al. (2005) with minor modifications. The retrotransposon-based primer C09-gypsy + C (Syed et al. 2006) in combination with each of 9 MseI primers was analysed in a pre-screening using a subset of five samples. The MseI primers M38 (ACT) and M62 (CTT) were selected for analysis of the total sample based on clarity of the electrophoresis profiles and level of polymorphism observed. The separation, visualisation and scoring of fragments followed the procedures performed for AFLPs.
SRAP analyses were carried out according to the procedures of Ferriol et al. (2003) with slight modifications. A pre-screening, as performed for SSAP, was carried out for the forward primers me1, me2 and me3 in combination with each of the reverse primers em1, em2 and em3. Details on SRAP primers are described in Li and Quiros (2001). The total sample was analysed for primer me2 in combination with each of the primers em2 and em3. Prior to PCR, the forward primer was radiolabelled with P33. Separation, visualisation and scoring of fragments were as described for AFLPs.
TRAP analysis basically followed the procedures of Hu and Vick (2003). Four fixed primers based on lettuce ESTs, QGA7H07L, QGA7H07R, GGB9J18L and GGB9J18R in combination with each of the arbitrary primers Ga3-800 and Sa4-700 (Hu et al. 2005) were tested in a pre-screening, as performed for SSAP. The arbitrary primers were radiolabelled with P33 prior to PCR. The primer combinations QGA7H07L/Sa4-700, QGA7H07R/Ga3-800, GGB9J18L/Sa4-700 and GGB9J18R/Ga3-800 were used to screen the total sample. The methods described for AFLP were used to separate, visualise and score TRAP fragments.
NBS-analyses were performed according to the procedures described by van der Linden et al. (2004). The MseI adapter primer was combined with each of the NBS anchored primers NBS3 and NBS5A (Syed et al. 2006) to screen the total sample. NBS specific primers were radiolabelled with P33 prior to PCR, and NBS fragments were separated, visualised and scored as performed for AFLPs.
Morphological characterisation
Seeds were sown in a greenhouse in March 2007, and 45 plants per accession were transferred in April to an experimental field location in Wageningen, The Netherlands. Plants were arranged in three rows with 30 cm inter-plant distance. Accessions were scored for 27 characters (Table 2), largely resembling the descriptor list presented by Boukema et al. (1990), and including nearly all minimum descriptors developed for cultivated lettuce and wild Lactuca spp. (Thomas et al. 2005).
Because the morphological data included quantitatively and qualitatively scored characters, and because the scale of scoring varied among the descriptors, a similarity matrix was constructed as follows. Per character, pair-wise differences between scores were calculated for all samples. Subsequently, these differences were standardised by subtracting the mean and dividing by the standard deviation of all pair-wise comparisons. The average standardised difference was then calculated over all characters for each pair-wise comparison, and transformed to similarity values on a scale from zero to one.
Expert-based qualifications
To obtain a reference population structure for the investigated samples, three crop experts with a long-lasting experience with CGN’s lettuce collection were consulted, namely a lettuce breeder affiliated to the breeding company Nunhems Netherlands BV, the former curator of CGNs lettuce collection and the former representative from The Netherlands Inspection Service for Horticulture responsible for lettuce variety registration. The experts were presented a list with the 80 investigated accessions accompanied with all relevant passport data. Based on their knowledge about the accessions, the passport data and visual examination of the material during the experimental field trial, expert-based similarities were estimated for each pair of accessions. To avoid that experts had to fill in a 80 × 80 diagonal matrix, the experts were requested to fill in a 10 × 10 diagonal matrix for each of the eight subgroups and a 8 × 8 diagonal matrix for the pair-wise comparison of all subgroups. These matrices were combined into a single similarity matrix for each expert and subsequently used to perform UPGMA cluster analyses. The datasets were modified until the resulting dendrograms adequately represented the experts’ view about the genetic relationships among the accessions. The three final 80 × 80 matrices were then used for further analysis. The experts worked independently from each other during all procedures.
Data analyses
Except for the co-dominantly scored microsatellites, fragments of different molecular weight were assumed to represent different loci, each having an allele for band presence and one for band absence. Homozygosity was assumed for all dominant marker scores. This assumption was considered sound because both L. sativa and L. serriola are known to exhibit predominantly self-fertilisation and because only a single case of heterozygosity was observed among all microsatellite data (0.12%). To express molecular diversity among accessions within groups (Table 1), gene diversity, or expected heterozygosity based on Hardy-Weinberg equilibrium (He), was calculated as 1 − Σp 2 i , where p i represents the frequency of the ith allele (Nei 1987). Molecular marker scores were also used to calculate the similarity of all pairs of samples according to the methods of Jaccard (1908). For this purpose, the microsatellite data were first transformed to binary scores. To estimate the quality of the marker datasets with respect to the ability to describe the genetic structure within the study material, data resolution (DR) values were calculated according to the methods described by van Hintum (2007).
To visualise genetic relationships among the accessions based on similarity values, UPGMA (unweighted pair group mean with arithmetic averaging) cluster analyses were carried out using NTSYS-pc (Rohlf 1993). Mantel tests (Mantel 1967) were performed to examine the degree of association between similarity matrices and to calculate Pearson’s correlation coefficient using GenStat (10th ed, VSN International Ltd, Oxford, UK). Correlation coefficients were tested for significant deviation from zero with a Student t test, where t = r√((n − 2)/(1 − r 2)), and r and n represent the correlation coefficient and the number of pair-wise comparisons, respectively (Sokal and Rohlf 1981). NTSYS-pc was used to visualise the degree of correlation among the similarity matrices generated on the basis of different characterisation methods by principal coordinates (PCO). For this purpose, the NTSYS module Dcenter was used to transform correlation coefficients to scalar products that subsequently were used to compute eigenvalues and eigenvectors with the NTSYS module Eigen.
Results
Variation among experts
The similarity matrices based on the individual experts’ assessments were significantly correlated. When examining the total sample of 80 accessions, correlation coefficients of 0.43, 0.57 and 0.65 were observed, whereas the correlation coefficient was never below 0.33 when separate analyses were performed for each of the seven main lettuce crop types and L. serriola. When the expert data were examined in relation to the marker data, the assessments of the individual experts were found to be complementary as the “performance” of the experts varied among the different subgroups (Fig. 1a). For example, expert 3 performed relatively well for butterhead lettuce, but rather poor for Latin lettuce in comparison with the other experts. Compared to individual assessments for the total sample, correlations with the marker data generally increased when the expert data were combined by averaging the values of the three similarity matrices (Fig. 1b). To facilitate the analyses, average similarity values of the experts were used in all further investigations.

Pearson’s correlation coefficient between expert-based similarities and a average similarity values of the different characterisation methods for each of the different subgroups, and b similarities of each of the different characterisation methods using the total sample
Estimated diversity
The number of scored polymorphic loci ranged from 11 for microsatellites to 275 for TRAP (Table 3). Because of the codominant nature of microsatellites, gene diversity estimates for SSRs, with a total of 142 alleles observed, were considerably higher compared to the dominantly scored markers. However, the DR of the SSR data set was only 0.49, which was considerably lower compared to the range of 0.80–0.91 observed for the other marker systems (Table 3). This indicated a rather poor data quality and implied that a higher number of SSR markers are required to achieve a DR value comparable to the other marker systems, and thus a comparable ability to describe the genetic structure of the study material.
In general, consistent gene diversities within groups were observed for the dominant markers. Compared with butterhead, cos, crisphead, cutting and Latin lettuce, gene diversity estimates were low in oilseed and stalk lettuce, while L. serriola showed relatively high values (Table 3a). Gene diversity estimates were generally in line with the average similarity values (Table 3b). Spearman’s rank correlation coefficient of the expert-based similarity values with the average molecular similarities was 0.45, whereas that with the morphology-based similarities was only 0.07. This indicated that the experts did not base their qualifications solely on the visual examination of the material during the experimental field trial, but also on background knowledge of the accessions.
Population genetic structure
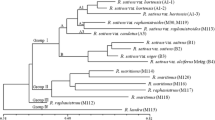
An UPGMA cluster analysis carried out for the expert-based similarities grouped the accessions according to species denomination and crop type classification within L. sativa (Fig. 2a). With respect to the distinction between L. sativa and L. serriola, and the clustering of accessions within each of the crop types oilseed and stalk lettuce, dendrograms constructed for individual markers roughly resembled the expert-based population genetic structure (results not shown). For the purpose of comparison, a population genetic structure based on the total set of molecular markers is shown in Fig. 2b. Although some clustering of accessions of the same crop type can be observed for butterhead, cos, crisphead, cutting and Latin lettuce, distinction of these crop types was much less evident compared with the expert-based dendrogram. In the molecular dendrogram, CGN10975 clustered with two cos accessions outside its supposed crop type group oilseed lettuce, while the cos accession CGN04628 clustered within the group of stalk lettuces. During the morphological field trial, accession CGN10975 was reclassified as cos lettuce, while accession CGN04628 showed characteristics of both cos and stalk. Accession CGN09356 was also reclassified as cos lettuce but, despite its rather large genetic distance with the other group members, it clustered within oilseed lettuce. CGN20693 was redetermined as L. saligna, and appeared as the most distinct accession within the L. serriola group in the expert-based as well as the marker-based dendrogram.


UPGMA cluster analysis of the accessions performed for a the average expert-based similarities, and b the average molecular marker-based similarities. Accessions are displayed by their CGN accession identifier, preceded by their registered subgroup classification (But butterhead, Cos cos, Lat Latin, Cut cutting, Cri crisphead, Sta stalk, Oil oilseed, Lse L. serriola). Groups of accessions that clustered according to subgroup classification are indicated in the right margin
Correlations between characterisation data
When the total sample was considered, similarity values of the different molecular marker systems, the morphological description and the expert’s assessments were all significantly correlated. These strong correlations were largely due to the clear distinction between L. sativa and L. serriola that was easily identified by all characterisation methods. This indicated that the choice of characterisation method is relatively unimportant to discriminate germplasm at higher taxonomic level.
In order to evaluate correlations between the different characterisation methods at lower taxonomic levels, separate analyses were carried out for each of the main lettuce crop types and L. serriola. In general, the morphological data showed a low level of association with the molecular data as they occupied opposite positions in the PCO plots for cos, crisphead, cutting and stalk lettuce and L. serrriola (Fig. 3), suggesting that a different kind of diversity is measured by the two characterisation methods. However, compared with the molecular data, the morphological data did not show a consistently better relationship with the experts’ assessments (Table 4).

Principal co-ordinate plots illustrating the correlation between the characterisation methods for each of the main crop types of cultivated lettuce and L. serriola (see Material and methods for details on methodology). The percentage of variation explained by each of the two principal axes is denoted in parentheses in the axis legend
Because random processes are expected to affect all loci simultaneously and to the same extent, a higher degree of correlation may be expected among the anonymous markers microsatellites, AFLP and SSAP as compared to that among the targeted markers SRAP, TRAP and NBS. However, tighter clustering of anonymous markers was observed for none of the groups presented in Fig. 3. The PCO plots did neither reveal a consistent pattern of the expert data being more strongly correlated with the targeted marker data than with the anonymous marker data. None of the molecular markers were significantly correlated with the experts’ assessments for all the investigated groups, nor did any of the markers consistently show the highest correlation with the experts across all groups (Table 4). The number of non-significant correlations with the expert data ranged from one for TRAP to four for NBS. TRAP markers were uncorrelated with the expert data only for Latin lettuce, but for this crop type all marker systems performed rather poor with the exception of AFLPs. In summary, our analyses did not indicate a substantial better performance of the targeted markers, either overall or individually, as compared to the anonymous markers.
Discussion
Comparison of markers
In our study, we compared anonymous and targeted molecular marker systems for their ability to describe the genetic diversity in a selected set of Lactuca accessions. In order to facilitate this comparison, a population structure was needed that could be used as a reference. For this purpose, the expertise of three crop experts was used. Similarity matrices based on the experts’ assessments were found to be significantly correlated and to be complementary to each other. The additive nature of the expertise of crop experts was also demonstrated in a study on the methodology for the selection of material to add to the CGN lettuce collection (van Treuren et al. 2008). Combining the experts’ data into a single similarity matrix generally outperformed the individual assessments, and, therefore, the use of a single reference genetic structure was considered a sound procedure.
Morphological data appeared only weakly associated with the molecular data in our study. Moreover, compared with the molecular data, no consistently better relationships were observed with the experts’ assessments. Although most genebanks invest considerable efforts in the morphological characterisation of germplasm (Engels and Visser 2003), its usefulness for the estimation of diversity in the collection appeared limited in our study. Limited sets of morphological characters seem more appropriate for verification purposes, such as the validation of the taxonomic status of material at the level of the species, subspecies and crop type, rather than for the adequate estimation of genetic relationships at low taxonomic levels, such as among cultivars within lettuce crop types in the present study. Moreover, morphological field trials are of interest to users of genetic resources because of the opportunity to examine the general performance and characteristics of material that could be potentially interesting for breeding purposes.
No substantial differences in performance were observed between the targeted and anonymous molecular markers in our study. Several factors underlying this finding may be considered. First, an effect of marker system may be obscured by methodological bias resulting from poor data quality due to a limited number of studied loci and/or populations. Estimation of DR values offers the opportunity to quantify the quality of data sets (van Hintum 2007). With the exception of microsatellites, DR-values of the molecular marker systems appeared relatively large and of similar magnitude. It was, therefore, considered unlikely that the similarity in results between the marker systems was due to methodological bias. Compared to dominant markers, such as AFLPs, considerably lower numbers of codominant loci, such as microsatellites, are usually studied to estimate genetic diversity. The lower DR-value of microsatellites indicated that a higher number of loci would be needed to reach a data resolution level similar to that observed for the other marker systems. Also in a simulation study using microsatellite and AFLP markers, it was shown that with increasing levels of genomic heterogeneity between loci, higher numbers of microsatellite loci are needed to predict whole genome diversity and that AFLPs are to be preferred in that case (Mariette et al. 2002). Second, the fraction of selective loci may be small among targeted markers, which may cause them to “behave” largely like anonymous markers. Although matching of amplified fragments with the target sequences has been demonstrated in SRAP (Li and Quiros 2001) and NBS (Syed et al. 2006), the extent to which this applied to the markers in our study was not examined. Moreover, variation in a target sequence does not necessarily imply a phenotypic effect. It, therefore, remained unclear which part of the observed variation for targeted markers has been affected by selection. Third, a clear and prominent population structure may be present that is easily revealed, irrespective of the marker system used. This was observed when the total sample was analysed, and all similarity matrices were significantly correlated due to the clear distinction between L. sativa and L. serriola. However, the population structure within L. sativa was found to be less clear-cut, as shown by the differences between the expert-based and molecular marker-based dendrogram in Fig. 2. Although evolutionary trees are generally considered useful to describe population genetic structure, their accuracy has been shown to depend on the method of tree construction and the evolutionary history of the populations. Particularly in case of complex genetic population structures, UPGMA trees should be interpreted with some caution (Kalinowski 2009).
For oilseed lettuce, low levels of variation and high correlations among all diversity matrices were observed. All oilseed accessions at CGN originated from Egypt and may have been derived from small founding events. If, in general, the degree of correlation among markers increases with higher levels of homogeneity, correlating markers to phenetic groups may even be more problematic when dealing with highly heterogeneous material, such as in the case of outbreeders.
Lettuce diversity
Cultivated lettuce is generally considered to have domesticated in the Middle East, with L. serriola being at least one of its direct ancestors and oilseed and stalk lettuce representing primitive forms. Cos lettuce is thought to have evolved from stalk lettuce and to have provided for a rich source of diversity for the development of butterhead, crisphead, cutting and Latin lettuce (Lebeda et al. 2007; Mou 2008). This history of cultivation was to a large extent reflected in the expert-based as well as the molecular marker based dendrogram (Fig. 2) as both showed an intermediate position of the primitive crop types between L. serriola and the other crop types.
For the butterhead, crisphead, cutting, cos and Latin lettuces, the clustering of accessions according to crop type classification was much less evident in the molecular marker-based dendrogram as compared to that of the crop experts. This limited molecular genetic structure for the modern crop types may be related to the intensive breeding activities employed in the development of new lettuce varieties. For example, in the European Common Catalogue (http://ec.europa.eu/food/plant/propagation/catalogues/index_en.htm), as many as 679 lettuce varieties with documented crop type were introduced during the period 2002 up and till 2006, comprising 243 butterhead, 183 crisphead, 167 cutting, 58 cos and 28 Latin lettuces. Breeding programs in lettuce are largely directed to resistance to pest and diseases, for which the crop-related wild gene pool is often exploited (Lebeda et al. 2007; Mou 2008). In addition, crossing barriers do not exist among different crop types and often breeding parents of different crop type are used in the development of new varieties (Hu et al. 2005). This practice of lettuce breeding may have blurred the boundaries between the crop type gene pools, resulting in reduced genetic structure. Previously, microsatellite screening of 414 lettuce varieties registered in the Dutch section of the European Common Catalogue during the period 1997–2001 demonstrated overlap between different crop types and the occurrence of intermediate types (van Treuren et al. 2008). Screening of 53 lettuce cultivars with 388 polymorphic TRAP markers also showed exceptions to the grouping of varieties by crop type (Hu et al. 2005). Similar results were obtained from the screening of 90 lettuce cultivars with 61 EST-SSRs (Simko 2008). In the latter study, a mixed cluster of butterhead and Latin lettuces was observed, a result also found in our study (Fig. 2b top cluster). These results suggest that apart from a number of distinct characters associated with crop type, overall genetic relationships among lettuce varieties belonging to the same registered crop type are more complex.
Concluding remarks
It can be argued that genomic variation that is, or might become, of adaptive significance should be the diversity to optimise in crop germplasm collections. However, the question is which of the currently available characterisation methods can be considered the most appropriate to estimate this variation. The method of choice should ideally generate information from many functional genomic regions that collectively constitute a representative sample from the total expressed DNA. For the purpose of germplasm characterisation and diversity estimation, no added value was observed in the present study for the targeted molecular markers SRAP, TRAP and NBS as compared with the anonymous markers microsatellites, AFLP and SSAP. First, markers targeted to specific gene sequences may still behave as anonymous markers, unless the relationship between molecular and phenotypic variation has been established. Secondly, the type of marker system used is irrelevant when at low taxonomic levels a clear genetic structure is absent due to intensive breeding activities.
References
Boukema IW, Hazekamp Th, van Hintum ThJL (1990) The CGN collection reviews: the CGN lettuce collection. Centre for Genetic Resources, The Netherlands (CGN), Wageningen
Bretting PK, Widrlechner MP (1995) Genetic markers and plant genetic resource management. Plant Breed Rev 13:11–86
Brown SM, Kresovich S (1996) Molecular characterization for plant genetic resources conservation. In: Paterson H (ed) Genome mapping of plants. Academic Press, San Diego, pp 85–93
Brugmans B, Fernandez del Carmen A, Bachem CWB, van Os H, van Eck HJ, Visser RGF (2002) A novel method for the construction of genome wide transcriptome maps. Plant J 31:211–222
Budak H, Shearman RC, Parmaksiz I, Gaussoin RE, Riordan TP, Dweikat I (2004) Molecular characterization of Buffalograss germplasm using sequence-related amplified polymorphism markers. Theor Appl Genet 108:328–334
Engels JMM, Visser L (2003) A guide to effective management of germplasm collections. IPGRI handbook for genebanks No. 6. International Plant Genetic Resources Institute, Rome
Falconer DS (1981) Introduction to quantitative genetics, 2nd edn. Longman, London
Ferriol M, Picó B, Nuez F (2003) Genetic diversity of a germplasm collection of Cucurbita pepo using SRAP and AFLP markers. Theor Appl Genet 107:271–282
Frankel OH, Brown AHD (1984) Plant genetic resources today: a critical appraisal. In: Holden JHW, Williams JT (eds) Crop genetic resources: conservation and evaluation. George Allen & Urwin Ltd, London, pp 249–257
Fulton TM, Chunwongse J, Tanksley SD (1995) Microprep protocol for extraction of DNA from tomato and other herbaceous plants. Plant Mol Biol Rep 13:207–209
Graner A, Streng S, Kellermann A, Schiemann A, Bauer E, Waugh R, Pellio B, Ordon F (1999) Molecular mapping and genetic fine-structure of the rym5 locus encoding resistance to different strains of the Barley Yellow Mosaic Virus Complex. Theor Appl Genet 98:285–290
Hu J, Vick BA (2003) Target region amplification polymorphism: a novel marker technique for plant genotyping. Plant Mol Biol Rep 21:289–294
Hu J, Ochoa OE, Truco MJ, Vick BA (2005) Application of the TRAP technique to lettuce (Lactuca sativa L.) genotyping. Euphytica 144:225–235
Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bulletin Société Vaudoise Sciences Naturelles 44:223–270
Jeuken M, van Wijk R, Peleman J, Lindhout P (2001) An integrated interspecific AFLP map of lettuce (Lactuca) based on two L. sativa × L. saligna F2 populations. Theor Appl Genet 103:638–647
Kalinowski ST (2009) How well do evolutionary trees describe genetic relationships among populations? Heredity 102:506–513
Karp A, Kresovich S, Bhat KV, Ayad WG, Hodgkin T (1997) Molecular tools in plant genetic resources conservation: a guide to the technologies. IPGRI technical bulletin No. 2. International Plant Genetic Resources Institute, Rome
Křístková E, Doležalová I, Lebeda A, Vinter V, Novotná A (2008) Description of morphological characters of lettuce (Lactuca sativa L.) genetic resources. Hort Sci (Prague) 35(11):3–129
Lebeda A, Ryder EJ, Grube R, Doležalová I, Křístková E (2007) Lettuce (Asteraceae; Lactuca spp.). In: Singh RJ (ed) Genetic resources, chromosome engineering, and crop improvement, vegetable crops, vol 3. CRC Press, Boca Raton, pp 377–472
Li G, Quiros CF (2001) Sequence-related amplified polymorphism (SRAP), a new marker system based on a simple PCR reaction: its application to mapping and gene tagging in Brassica. Theor Appl Genet 103:455–461
Mantel N (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Mariette S, Le Corre V, Austerlitz F, Kremer A (2002) Sampling within the geneome for measuring within-population diversity: trade-offs between markers. Mol Ecol 11:1145–1156
Mou B (2008) Lettuce. In: Prohens J, Nuez F (eds) Handbook of plant breeding, vegetables I, Asteraceae, Brassicaceae, Chenopodicaceae, and Cucurbitaceae, vol I. Springer, New York, pp 75–116
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Queller DC, Strassmann JE, Hughes CR (1993) Microsatellites and kinship. Trends Ecol Evol 8:285–288
Reed DH, Frankham R (2001) How closely correlated are molecular and quantitative measures of genetic variation? A meta-analysis. Evol Int J Org Evol 55:1095–1103
Rohlf FJ (1993) NTSYSpc numerical taxonomy and multivariate analysis system, Version 2.1. Exeter Software, New York
Simko I (2008) Development of EST-SSR markers for the study of population structure in lettuce (Lactuca sativa L.). J Hered. doi:10.1093/jhered/esn072
Sokal RR, Rohlf FJ (1981) Biometry, 2nd edn. WH Freeman and Company, United States
Spooner D, van Treuren R, de Vicente MC (2005) Molecular markers for genebank management. IPGRI technical bulletin No. 10. International Plant Genetic Resources Institute, Rome
Syed NH, Sureshsundar S, Wilkinson MJ, Bhau BS, Cavalcanti JJV, Flavell AJ (2005) Ty1-copia retrotransposon-based SSAP marker development in Cashew (Anacardium occidentale L.). Theor Appl Genet 110:1195–1202
Syed NH, Sørensen AP, Antonise R, van de Wiel CCM, van der Linden G, van’t Westende W, Hooftman DAP, den Nijs HCM, Flavell AJ (2006) A detailed linkage map of lettuce base don SSAP, AFLP and NBS markers. Theor Appl Genet 112:517–527
Thomas G, Astley D, Boukema IW, Daunay MC, Del Greco A, Díez MJ, van Dooijeweert W, Keller J, Kotlińska T, Lebeda A, Lipman E, Maggioni L, Rosa E, compilers (2005) Report of a vegetables network. Joint Meeting with an ad hoc group on leafy vegetables, 22–24 May 2003, Skierniewice, Poland. International Plant Genetic Resources Institute, Rome
van de Wiel CCM, Arens P, Vosman B (1999) Microsatellite retrieval in lettuce (Lactuca sativa L. Genome 42:139–149
van der Linden CG, Wouters DCAE, Mihalka V, Kochieva EZ, Smulders MJM, Vosman B (2004) Efficient targeting of plant disease resistance loci using NBS profiling. Theor Appl Genet 109:384–393
van Hintum ThJL (1999) The core selector, a system to generate representative selections of germplasm accessions. Plant Genet Res Newsl 118:64–67
van Hintum ThJL (2007) Data resolution: a jackknife procedure for determining the consistency of molecular marker datasets. Theor Appl Genet 115:343–349
van Treuren R, van Hintum ThJL, van de Wiel CCM (2008) Marker-assisted optimization of an expert-based strategy for the acquisition of modern lettuce varieties to improve a genebank collection. Genet Res Crop Evol 55:319–330
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucl Acid Res 23:4407–4414
Waugh R, McLean K, Flavell AJ, Pearce SR, Kumar A, Thomas BBT, Powell W (1997) Genetic distribution of Bare-1-like retrotransposable elements in the barley genome revealed by sequence-specific amplification polymorphisms (S-SAP). Mol Gen Genet 253:687–694
Acknowledgments
The authors would like to thank Aad van der Arend, Ietje Boukema, Nico van Marrewijk and Sebo van der Wal for their contributions in assessing the reference population structures, and Hanneke van der Schoot, Yolanda Noordijk and Wendy van’t Westende for carrying out the molecular analyses. They are also grateful to Dr. C. F. Quiros for proving information about suitable SRAP primer combinations, to Liesbeth de Groot for her involvement in the field experiments, and to two anonymous referees for their comments on an earlier version of the paper. This project was partly financed by the Dutch Ministry of Agriculture, Nature and Food Quality. The morphological examinations were part of a more comprehensive characterisation trial carried out within the framework of the EU-funded GENRES project ‘Leafy Vegetables germplasm, stimulating use’ (AGRI-2006-0262).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Graner.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
van Treuren, R., van Hintum, T.J.L. Comparison of anonymous and targeted molecular markers for the estimation of genetic diversity in ex situ conserved Lactuca . Theor Appl Genet 119, 1265–1279 (2009). https://doi.org/10.1007/s00122-009-1131-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-009-1131-1