
Abstract
A unification of doping-control screening procedures of prohibited small molecule substances—including stimulants, narcotics, steroids, β2-agonists and diuretics—is highly urgent in order to free resources for new classes such as banned proteins. Conceptually this may be achieved by the use of a combination of one gas chromatography–time-of-flight mass spectrometry method and one liquid chromatography–time-of-flight mass spectrometry method. In this work a quantitative screening method using high-resolution liquid chromatography in combination with accurate-mass time-of-flight mass spectrometry was developed and validated for determination of glucocorticosteroids, β2-agonists, thiazide diuretics, and narcotics and stimulants in urine. To enable the simultaneous isolation of all the compounds of interest and the necessary purification of the resulting extracts, a generic extraction and hydrolysis procedure was combined with a solid-phase extraction modified for these groups of compounds. All 56 compounds are determined using positive electrospray ionisation with the exception of the thiazide diuretics for which the best sensitivity was obtained by using negative electrospray ionisation. The results show that, with the exception of clenhexyl, procaterol, and reproterol, all compounds can be detected below the respective minimum required performance level and the results for linearity, repeatability, within-lab reproducibility, and accuracy show that the method can be used for quantitative screening. If qualitative screening is sufficient the instrumental analysis may be limited to positive ionisation, because all analytes including the thiazides can be detected at the respective minimum required levels in the positive mode. The results show that the application of accurate-mass time-of-flight mass spectrometry in combination with generic extraction and purification procedures is suitable for unification and expansion of the window of screening methods of doping laboratories. Moreover, the full-scan accurate-mass data sets obtained still allow retrospective examination for emerging doping agents, without re-analyzing the samples.
Similar content being viewed by others


Introduction
The task of doping control laboratories is to screen for a wide range of drugs currently included in the list of prohibited substances published by the World Anti-Doping Agency (WADA) [1]. Today, the cost-effectiveness of analytical procedures is becoming an important issue for all laboratories involved in doping control or residue analysis. A way to improve cost-effectiveness is to maximize the number of analytes that may be determined in a single procedure, e.g. to use multi-compound techniques. However, most methods are developed for the determination of specific prohibited compounds or compound groups. Because resources are needed to determine new classes of doping compounds such as banned proteins, unification of doping control screening procedures for prohibited small molecule substances—including stimulants, narcotics, steroids, β2-agonists, and diuretics—is highly urgent. Conceptually this may be achieved by the use of a combination of one gas chromatography–time-of-flight mass spectrometry (GC–TOFMS) method and one liquid chromatography time-of-flight mass spectrometry (LC–TOFMS) method. The aim of this study is to develop and validate a multi-compound LC–TOFMS method for compounds that are less suitable for GC–MS methods. Traditionally, capillary gas chromatography combined with mass spectrometry (GC–MS) has been used in doping analysis including laborious sample preparation for analytes or metabolites with a polar, non-volatile, or thermolabile nature [2]. As an alternative, liquid chromatography combined with mass spectrometry (LC–MS) may be used for those targeted molecules in urine that cannot be covered by standard GC–MS methods. While LC combined with tandem mass spectrometry (LC–MS–MS) has excellent sensitivity and selectivity for target analytes in doping analysis [3–6], true multi-compound analyses requires a sensitive full-scan MS technique, for example time-of-flight mass spectrometry (TOFMS) or Orbitrap mass spectrometry [7–9]. These analyzers provide high specificity because of both high mass accuracy and high mass resolution and allow the reconstruction of highly selective accurate mass chromatograms for a theoretically unlimited number of compounds in complex matrices. Furthermore, data can be acquired and reprocessed without any a priori knowledge about the presence of specific compounds; that is, no analyte-specific information is required before injecting a sample and the presence of newly identified compounds can be confirmed in previously analysed samples simply by reprocessing the data. The advantage of TOFMS can be further improved by combining it with high-resolution LC (HRLC) such as ultra performance LC (UPLC). Recent research has shown that UPLC–TOFMS has significant advantages concerning selectivity, sensitivity, and speed [10–14]. This paper describes the development and validation of a quantitative screening method based on UPLC–TOFMS for analysis of 56 restricted substances that are not very suitable for detection with GC–MS methods, including corticosteroids, β2-agonists, and diuretics. Corticosteroids affect the nervous system causing euphoria, alleviate pain, and enhance the athlete’s ability to concentrate in endurance or power events [15, 16]. (Gluco)corticosteroids show extensive metabolism in the human body and are generally excreted in urine at low concentrations complicating the analysis of these compounds [17, 18]. Most corticosteroids are non-volatile, therefore excluding GC–MS analysis of urine samples unless lengthy derivatization or oxidation procedures are used prior to GC–MS analysis. Generally, LC–MS–MS with different ionization techniques has been used [19–21]. In addition LC–TOFMS has been used for toxicological drug screening of related compounds [22] and specifically for doping agents in human urine [23]. The minimum required performance limit (MRPL) established for these compounds, at which all laboratories should be able to perform [24], is 30 ng mL−1 in urine. The structures and names of the 19 corticosteroids involved in this study are given in Fig. 1.

(Cortico)steroids involved in this study: budesonide (a), prednisolone (b), prednisone (c), desonide (d), methylprednisolone (e), cortisone (f), cortisol (g), flumethasone (h), flucortisone (i), fluocortolone (j), fluniside (k), triamcinolone acetonide (l), dexamethasone (m), betamethasone (n), triamcinolone (o), gestrinone (p), tetrahydrogestrinone (THG) (q), trenbolone (r), epitrenbolone (s)
β2-Agonists act on the β2 adrenergic receptor, causing muscle relaxation resulting in a widening of the bronchial passages and blood vessels in the muscles, thereby improving the performance of the athletes [25]. β2-Agonists can be divided into short-acting and long-acting groups with salbutamol and terbutaline as examples of the first group and salmeterol and clenbuterol as examples of the latter group. β2-Agonists, for example salbutamol, have become a concern in sports, because these drugs, when used at high doses, can act as anabolic agents to promote weight, mainly in the form of muscle [26]. As of 2006, WADA permits the use of selected β2-agonists (salbutamol, salmeterol, terbutaline, and eformoterol) in athletic competition only by asthmatic athletics. For salbutamol the threshold value is 1000 ng mL−1 while for all other β2-agonists the MRPL is 100 ng mL−1. β2-Agonists in urine have been analysed using LC–MS–MS [27] and LC–TOFMS [23], but also LC combined with Orbitrap mass spectrometry [28], in all cases after hydrolysis and liquid–liquid extraction of the sample. The structures and names of the 22 β2-agonists involved in this validation study are given in Fig. 2.

β2-Agonists involved in this study: clenbuterol (a), clenproperol (b), clenpenterol (c), clencyclohexerol (d), brombuterol (e), mapenterol (f), salbutamol (g), cimaterol (h), cimbuterol (i), mabuterol (j), salmeterol (k), zilpaterol (l), carbuterol (m), terbutaline (n), clenhexyl (o), isoxsuprine (p), procaterol (q), fenoterol (r), ractopamine (s), tulobuterol (t), reproterol (u), formoterol (v)
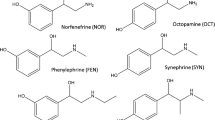
Diuretics increase the urine flow, thereby reducing body mass and potentially leading to the athlete’s classification in a lower weight class in some sports. Thiazide diuretics are also regarded as masking agents, because the increased urine flow and volume dilute possible residues of sports doping and for these reasons all diuretics appear on the prohibited list, both in and out of competition with an MRPL of 250 ng mL−1. Diuretics have been determined as their methyl derivatives by GC–MS [29, 30] and by liquid chromatography combined with tandem mass spectrometry (LC–MS–MS) [31–33]. The latter option does not require derivatization and enables simplified sample preparation because of to the compatibility of aqueous samples with the analytical system. Figure 3 gives the structures and names of the thiazide diuretics involved in this study. Narcotics and stimulants can be misused in sports and are therefore regarded as doping agents by WADA with MRPL values of 200 ng mL−1 for narcotics and 500 ng mL−1 for stimulants. Narcotics and stimulants have been analysed in urine using GC–MS [34], often in combination with solid-phase micro extraction (SPME) [35, 36], and, incidentally, using LC–MS–MS [37]. In this study only a few narcotics and stimulants are considered and their structures and names are given in Fig. 4. Doping analysis has to comply with the requirements of International Standards for Laboratories established by WADA, including a chain of custody, validation of screening and confirmation methods, and criteria for identification [38]. Therefore, newly developed methods have to be validated before they can be used in official control studies to demonstrate that the specific requirements are met by the analytical method. The WADA international standard for laboratories describes which method performance data should be determined, however, without stating a procedure for their determination. In this study a validation procedure is used based on EC commission decision 2002/657/EC, as used by many EU laboratories involved in residue analysis in food [39].

Thiazide diuretics involved in this study: chlorothiazide (a), hydrochlorothiazide (b), hydroflumethiazide (c), benzthiazide (d), bendroflumethiazide (e), althiazide (f), trichlormethiazide (g), methyclothiazide (h), cyclothiazide (i), polythiazide (j)

Narcotics and stimulants involved in this study; sydnocarb (a), oxycodone (b), phenylephrine (c), mephentermine (d), methoxyphenamine (e)
Materials and methods
All steroid reference substances were obtained from RIVM (Bilthoven, The Netherlands), Sigma (Zwijndrecht, The Netherlands), or Steraloids (Rhode Island, USA). Fluocortolone, the thiazide diuretics, and the narcotics and stimulants were a kind gift from the Doping Control Laboratory of Athens (OAKA, Greece). All steroid substances were obtained and handled in accordance with local legislation. Oasis MCX cartridges were obtained from Waters (Milford, MA, USA). All solvents were of HPLC-grade or higher. Acetonitrile, acetone, methanol, and water were purchased from Biosolve (Valkenswaard, The Netherlands). Sodium acetate, sodium (bi)carbonate, and leucine-enkephalin were purchased from Sigma. Ethyl acetate, formic acid, acetic acid and β-glucuronidase/arylsulfatase (from Helix pomatia) were purchased from Merck (Amsterdam, The Netherlands). All urine samples analysed within the validation procedure were spontaneous urine samples obtained from healthy male and female volunteers. To account for matrix effects samples were collected from seven volunteers resulting in a total of 21 samples for the three validation days. The samples were collected in glass bottles and, after homogenization, subdivided into smaller PE bottles and stored at −20 °C until use.
For sample preparation 3 mL urine was mixed with 2 mL sodium acetate buffer (0.25 mol L−1; pH 4.8) and shaken. The urine samples were subsequently enzymatically deconjugated for 1.5 h at 50 °C using 25 μL β-glucuronidase/arylsulfatase and left to cool to room temperature. An Oasis MCX cartridge (60 mg, 3 mL) was conditioned with 3 mL methanol followed by 3 mL sodium acetate buffer (0.25 mol L−1; pH 4.8) before applying the deconjugated urine samples. The cartridges were washed with 1 mL 1 mol L−1 acetic acid followed by 3 mL 15% acetone in sodium acetate buffer (0.25 mol L−1; pH 4.8). After drying by vacuum the compounds of interest were eluted using 3 mL 3% ammonia solution (as NH3) in ethyl acetate. The solvent was evaporated at 40 °C using a flow of nitrogen gas until just dry and the residue was reconstituted in 200 μL 95:5 water–acetonitrile. Separation of the sample was performed on a Waters Acquity UPLC system consisting of a vacuum degasser, an autosampler with a cooled sample tray, a column oven, and a binary solvent manager with high-pressure mixing chamber. Separation was performed at 35 °C using a Waters Acquity BEH-C18 column (100 × 2.1 mm i.d., 1.7 μm particle size). The eluents for both positive and negative electrospray ionisation consisted of 0.1% formic acid (A) and acetonitrile–0.1% formic acid, 9:1 (v/v) (B). Ultra pure, LC–MS-grade water from Biosolve (Valkenswaard, The Netherlands) was used to eliminate excessive background signals and avoid the formation of sodium or potassium adducts. A step-wise gradient starting at 0% B was employed at a flow of 0.4 mL min−1. From 1 to 4 min the %B was linearly increased to 40% and during 4 to 10 min linearly increased to 100% with a final hold for 2 min. The total run-to-run time (including equilibration prior to injection of the next sample) was 13 min. The injection volume was 20 μL.
The effluent from the UPLC system was directly interfaced to a Bruker Daltonics micrOTOF mass spectrometer equipped with an orthogonal electrospray ionisation (ESI) source operated in the positive (all compounds except thiazides) and negative (thiazides) modes using a mass range of m/z 100 to 1000. The trigger time was 33 μs and 10,000 spectra were summed, equalling 0.33 s time resolution. The capillary voltage of the ion source was set at 3500 V and the capillary exit at 100 V. The nebulizer gas pressure was 1.5 L min−1 and drying gas flow 8 L min−1. The drying temperature was set at 200 °C. Instrument calibration was performed externally before each sequence with a sodium formate–acetate solution using the theoretical exact masses of calibration ions with formula Na(HCO2Na)2–8 and Na(CH3CO2Na)2–8(HCO2Na)2–8 in the range m/z 100 to 1000 for calibration. Automated post-run internal mass scale calibration of individual samples was performed by injecting the calibrant at the beginning and end of each run via a six-port divert valve equipped with a 20-μL loop. The actual calibration was performed based on calibrant injection at the beginning of the run while the calibrant at the end of the run was for manual verification of calibration stability. The calibrator ions in the post-run internal mass scale calibration were the same as in the instrument calibration.
The developed method was validated in accordance with EU Commission Decision 2002/657/EC for a quantitative screening method, because this includes the WADA validation criteria and more. The validation study for the compounds in urine was carried out at three concentrations chosen around a validation level. This validation level (VL) was equal to the MRPL for corticosteroids and the threshold value for salbutamol, and to 0.5 times the MRPL for the other β2-agonists, thiazide diuretics, narcotics, and stimulants. The VL for the latter compound groups was set at 0.5 times the MRPL, because it was expected that that level could easily be determined by the LC–TOFMS method. Blank urine samples were fortified at 0.5, 1.0, and 2.0 times the VL level for all the target analytes and seven replicates of each concentration were analysed on one day. The 21 replicate analyses where repeated on two more days resulting in 63 independent determinations. Calibration curves were prepared from processed blank urine samples fortified with the target analytes, before instrumental analysis, at 0, 0.25, 0.5, 1.0, 2.0, and 4.0 times the VL levels. Each series of fortified samples on each of the three days started and ended with analysis of these matrix-matched calibration standards. From the data the repeatability, intra-laboratory reproducibility (both expressed as the relative standard deviation, RSD), and accuracy were calculated. The accuracy is expressed as the average recovery from samples at the VL level relative to a processed blank sample spiked before instrumental analysis. A range of 70–140% was considered acceptable for multi-compound quantitative screening as in this study. The linearity was determined for a concentration range of 0, 0.25, 0.5, 1, 2, and 4 times the VL level. On each validation day the calibration curves were constructed and the squared regression coefficients (r 2) calculated for each compound. Squared regression coefficients >0.99 were considered acceptable. The decision limit (CCα) and detection capability (CCβ) at the VL level were calculated from the standard deviation at the VL level using the following equations:
Note that TOFMS is a single MS system and is, therefore, according to EU regulation 2002/657/EC, by definition suitable for screening analysis only. This would exclude the determination of the decision limit CCα. However, because the WADA validation criteria do not pose such strict identification criteria for confirmation as the 2002/657/EC, a value for CCα also is included in this study. The robustness of the method was tested at the VL level by introduction of four small but deliberate changes in the operating procedure and by the assessment of their effect on the method results. These deliberate changes reflect those that may occur when a method is transferred between different laboratories. The effect of a particular variable was evaluated by comparison of the results from the deliberately modified method with those from the original method, taking into account the within-laboratory reproducibility. The specificity and selectivity of the method was checked by analysis of 20 representative blank samples and by the analysis of urine samples fortified with approximately 200 veterinary drugs and pesticides in addition to the target analytes at the VL level. The chromatograms of the blank samples were monitored for peaks that can potentially interfere with the analytes of interest while the results from the additionally fortified samples were compared with those from samples fortified with the target analytes only, taking into account the within-laboratory reproducibility. Finally, stability experiments were carried out for all analytes. Sample extracts were fortified at 0.5, 1.0, and 2.0 times the VL level and analysed after storage for four weeks at −20 °C. Because thiazide diuretics in urine have been reported to be unstable because of hydrolysis an additional experiment was carried out. Blank urine samples were spiked with thiazide diuretics at the VL level and analysed after storage at room temperature for 4 and 9 days.
Results
Generic sample preparation
A generic sample-preparation method was developed that was able to isolate and purify corticosteroids, β2-agonists, thiazide diuretics, and some additional narcotics and stimulants in one procedure from urine samples. The classical sample-preparation method consists of enzymatic hydrolysis of the urine sample using β-glucuronidase/aryl sulfatase, followed by extraction at pH 9 with diethyl ether. From preliminary experiments it became clear that while this method gives good results for corticosteroids, narcotics, and stimulants, the results for β2-agonists are poor because of interferences by matrix constituents. Additional sample purification was essential to eliminate these interferences and obtain spectra of sufficient quality to determine the correct accurate mass of the analytes of interest. However, a typical purification step as used for β2-agonists gave poor results for corticosteroids and thiazide diuretics, because most are lost during the washing step with chloroform. A combined extraction and clean-up procedure based on SPE was tested using an Oasis MCX cartridge, which is a polymeric mixed-mode cation-exchange–reversed-phase sorbent enabling the retention of acidic, neutral, and, especially, basic drugs. The SPE cartridge was conditioned using 3 mL methanol followed by 3 mL 0.25 mol L−1 sodium acetate (pH 4.8). Samples were applied to the cartridges, which were subsequently washed with either 3 mL water or 3 mL of an acetone–water mixture. Washing with water generally resulted in overestimation of the β2-agonists whereas washing with acetone–water (1:1) resulted in losses of the β2-agonists and to a lesser extent some of the thiazide diuretics. The alternative SPE method was further optimized using 10%, 15%, and 20% acetone in 0.25 mol L−1 sodium acetate to wash the cartridges. Finally, washing with 15% acetone in 0.25 mol L−1 sodium acetate at pH 4.8 was chosen as the best compromise. The analytes were eluted with 3 mL of an ammonia–ethyl acetate mixture. This alternative SPE method gives reasonable results for all compound groups.
High-resolution LC–TOFMS analysis
To develop the screening method, a solvent-based standard with all target analytes was analyzed in positive and negative modes. Based on the chromatographic retention times and the specific accurate masses and the isotope patterns calculated by the software from the elemental composition of the target analytes, a compound database is constructed. The isotope pattern-matching algorithm SigmaFit is a feature of the Bruker Daltonics micrOTOF that can be used as an identification tool in addition to accurate mass measurement. In the method the different combinations of retention time and accurate masses and the acceptable tolerances for these data and the SigmaFit value are defined. Following analysis of a real sample the full-scan chromatogram is processed and the target analytes are identified using the database and quantified using matrix-matched calibration standards. Chromatographic separation was performed with a C18 UPLC column and gradient elution. The repeatability of the analytes’ retention times was acceptable and the deviations from the expected retention times were generally were below 1%, with individual analytes up to 3% (Table 1). The latter was found for cortisone, an endogenous corticosteroid in urine with an often less symmetrical peak shape in the chromatograms, as illustrated by the extracted ion chromatogram (EIC) of cortisone in urine shown in Fig. 5. Figure 5 also shows the total ion chromatograms (TIC) of a blank urine sample (Fig. 5A), a solvent standard of the analytes (Fig. 5B), and a blank urine sample fortified at the 1.0 × VL level (Fig. 5C). At the beginning of the gradient the more hydrophilic analytes such as phenylephrine and the β2-agonist cimaterol and cimbuterol elute, while most of the corticosteroids elute in the region of 4 to 6 min. Peak shapes were generally good although asymmetrical peaks were observed for phenylephrine, probably because this peak elutes in the beginning of the gradient (Fig. 5D). For trenbolone two distinct peaks are observed for the diastereoisomers, 17β-trenbolone at 5.7 min and 17α-trenbolone at 5.9 min (Fig. 5J). Finally, the peak of tetrahydrogestrinone (THG) is a triplet, with the first peak being THG itself and the other two being matrix compounds that also show a response at the accurate mass of THG (313.2162) when a mass-tolerance window of 5 mDa is used (Fig. 5L). Even when the mass window is set to 2 mDa these peaks are present and only when a mass window of 1 mDa is used do these peaks disappear. Visually these peaks originate from the two peaks at 7.2 and 7.3 min that are clearly visible in TIC of the blank and the fortified urine samples in the chromatograms in Fig. 5A and C. The negative-ion mode data files are processed similarly and searched for the target analytes using Brukers Target Analysis software. Figure 6 shows the negative ion mode chromatograms of the blank urine sample (Fig. 6A), the solvent standard with all 56 added analytes (Fig. 6B), and the blank urine sample fortified with the analytes of interest at the 1.0 × VL level (Fig. 6C). Below, the extracted ion chromatograms of all thiazides with the exception of bendroflumethiazide are shown. Peaks and peak shapes are good with the exception of cyclothiazide (Fig. 6K) that shows a double peak. The structure of cyclothiazide shown in Fig. 3i contains four chiral centres and therefore 16 possible isomers. However, because of the restriction of the methylene bridgehead (cis only) the number of stereoisomers is reduced to eight, i.e. four diastereomeric racemates. Analysis of the standard material showed that this contained at least two partly separated isomers which explained the double peak. The results clearly show that all thiazides can be identified and quantified at the 1.0 × VL level.

Total ion and extracted ion chromatograms (TIC and EIC) of urine samples and individual analytes analysed in the positive mode. EICs of individual analytes are representative for all analytes and are extracted using a mass window of 5 mDa. From the top down the chromatograms shown are: TIC of a blank urine (A); TIC of a solvent standard of the analytes (B); TIC of a blank urine fortified at the 1.0 × VL level (C); EIC of phenylephrine (split peak just before 3 min), 30 μg L−1 (D); EIC of cimbuterol, 50 μg L−1 (E); EIC of clenbuterol, 2 μg L−1 (F); EIC of mapenterol, 50 μg L−1 (G); EIC of cortisone, >30 μg L−1 (H); EIC of beclomethasone, 30 μg L−1 (I); EIC of trenbolone, 30 μg L−1 (J); EIC of gestrinone, 30 μg L−1 (K); EIC of tetrahydrogestrinone (THG), 30 μg L−1 (L).

Total ion and extracted ion chromatograms (TIC and EIC) of urine samples and individual analytes analysed in negative-ion mode. EICs of individual compounds are representative for all analytes and are extracted using a mass window of 5 mDa. From the top down the chromatograms shown are: TIC of a blank urine (A); TIC of a solvent standard of the analytes (B); TIC of a blank urine fortified at 1.0 × the VL level (C); EIC of chlorothiazide, 100 μg L−1 (D); EIC of hydrochlorothiazide, 100 μg L−1 (E); EIC of hydroflumethiazide, 100 μg L−1 (F); EIC of trichloromethiazide, 100 μg L−1 (G); EIC of methyclothiazide, 100 μg L−1 (H); EIC of althiazide, 100 μg L−1 (I); EIC of benzthiazide, 100 μg L−1 (J); EIC of cyclothiazide, 100 μg L−1 (K); EIC of polythiazide, 100 μg L−1 (L)
The accurate mass measurement data obtained from spiked urine extracts are listed in Table 1. In general, for a TOFMS having a mass resolution of ∼10,000 FWHM and external calibration, a deviation of the measured accurate mass from the calculated mass of 10 ppm is acceptable, especially considering the sometimes low concentration levels [40]. In this study the average mass accuracy for individual compounds ranged from 1.1 to 5.8 ppm. However, for a few compounds mass measurement errors as high as 16 ppm were observed in individual measurements. The overall (all compounds in all measurements) average and median mass accuracy values were 2.8 and 2.6 ppm. These results are comparable with those of Ojanperä and Kolmonen, who both applied the same type of Bruker Daltonics micrOTOF system for analyses of drugs in urine and found mean mass measurement errors of 2.5 ppm and <5 ppm, respectively [22, 23]. The results are also comparable with mass measurement errors found in UPLC–TOFMS screening of veterinary drugs in food matrices, where average values of <10 and 3.0 ppm were found [10, 14]. An interesting observation is that the average mass measurement error decreases slightly with increasing concentration in accordance with a recent study by Bristow, who observed an increase of the deterioration in mass measurement accuracy at both very low and very high ion abundances [41].
A similar relationship is expected for the SigmaFit value, an exact numerical comparison of the theoretical and measured isotopic patterns of a target compound. The results indeed show that the SigmaFit value decreases with increasing concentration. This is according to expectations, because the signal-to-noise ratio increases with increasing concentration resulting in a better-defined isotope pattern. The SigmaFit value indicates the difference between theoretical and measured isotopic patterns, and low values (typically <0.1) indicate a good fit. This isotopic pattern match can be used as an orthogonal identification index together with the accurate mass and the retention time. In this study the average SigmaFit values for individual compounds range from 0.004 up to 0.53, and the difference between the average (0.109) and median (0.069) value indicates that there are only a few high SigmaFit values. These high SigmaFit values were found for cortisone, methylprednisolone, prednisolone, and prednisone, and to a lesser extent for reproterol, salbutamol, carbuterol, and cimaterol. With one exception, these compounds also show deviations of the expected retention time and calculated exact mass above average. For cortisone this is explained by the fact that this compound is endogenously present at concentrations much higher than the spiked concentrations. For methylprednisolone, prednisolone, and prednisone the observed deviations may be accounted for by the many matrix compounds eluting in this part of the chromatogram, and reproterol, carbuterol, and cimaterol elute at the beginning of the gradient which sometimes results in asymmetrical peak shapes. For salbutamol the reasons for these deviations are unknown. In general however, there seems to be a positive correlation between ΔRT, the mass error and the SigmaFit as visualized in Fig. 7, suggesting that co-elution of an isobaric matrix component occurs.

Deviations in RT (ΔRT), the mass error (Δm/z) and SigmaFit show some positive correlation
In-house method validation
The WADA international standard for laboratories [38] describes which method-performance characteristics should be determined, however, without stating a procedure for their determination. The required performance characteristics also depend on whether the method is intended for the determination of “non-threshold” or “threshold” substances. For non-threshold substances the method should be able to identify substances in the class of prohibited substances at the MRPL level but measurement of a concentration is not required. Because the results for non-threshold substances are not quantitative, criteria should be established to ensure that a substance representative of the class of prohibited substances can be repeatedly identified and detected as present in the sample at the MRPL level. This criterion is called the “identification capability” and is the same as the CCβ value in the well known EU document 2002/657/EC that defines and describes method validation for residue analysis in food [39]. For threshold substances the method should be capable of determining both, the identity and the concentration up and around the threshold level. Taking into account intermediate precision at the threshold level a “decision limit” is defined which is the same as the CCα value in the 2002/657/EC document. A good explanation of this concept can be found in the recent WADA technical document TD20009DL [42]. Because the WADA international standard for laboratories does not state a procedure for determination of the required performance data, the procedures in the 2002/657/EC document were followed and for reasons of clarity WADA’s identification capability and decision limit are also abbreviated as the CCβ and CCα values. Other validation data determined are precision/repeatability, reproducibility, accuracy/recovery, linearity, specificity, robustness and the stability of standards.
Table 2 presents the results for the performance characteristics of the method. No results are shown for cortisol and cortisone because both are endogenous compounds present in urine at concentrations far higher than the fortified concentrations and thus preventing reliable determination of the performance characteristics. There are also no results for clenhexyl, carbuterol, procaterol, and reproterol, because these could be identified in a limited number of samples only, not enabling us to calculate a reliable value for the performance characteristics. For all other compounds in this study the results in Table 2 show that the repeatability of the method ranges from 7 to 42% with an average value of 14%, and for 90% of the compounds the repeatability is <20%. The poorest repeatability is found for clenpenterol (42%) and zilpaterol (25%) as a result of limited sensitivity, and matrix interference. The intra-laboratory reproducibility ranges from 11 to 77% with an average value of 21%. The median value of 17% indicates that there are only a few poor reproducibility values and, as mentioned before, these are clenpenterol (77%) and zilpaterol (42%), but also salmeterol (67%). For 70% of the components the reproducibility is still <20%. In this study the validation level for the β2-agonists (and thiazide diuretics, narcotics, and stimulants) was set at 0.5 times the MRPL. As a consequence the actual repeatability and reproducibility at the MRPL level will most likely be better than the values presented in Table 2. The accuracy for the compounds in this study ranges from 68 to 143% with an average of 102% and a median value of 100% indicating a uniform distribution. The lowest accuracy was found for clenpenterol and the highest was found for carbuterol. In general it can be noticed that the accuracy for the individual corticosteroids, narcotics, and stimulants, and thiazide diuretics, are similar. On the other hand, much wider distribution is found for the accuracy of the individual β2-agonists which is explained by the washing step of the SPE column. Although a 15% acetone solution was finally selected it is clear that this remains a compromise that gives the best overall performance but results in individual differences. However, because the accuracy for 54 of the 56 compounds is in the range 70 to 140% we conclude that the method is capable of producing accurate screening results for all of the analytes with the exception of carbuterol and clenpenterol. The squared regression coefficient (r 2) is used to evaluate the linearity and is presented in Table 2. The matrix-matched calibration curves show r 2 > 0.99 for 90% of the analytes with exceptions for fluocortolone, salmeterol, and bendroflumethiazide. Although no limit of quantitation (LOQ) is actually determined, an indication of its value is estimated from the lowest calibration standard and is given in Table 2. The LOQ values range from 0.5 μg L−1 for compounds for which sensitivity is high, for example clenbuterol, to over 50 μg L−1 for compounds for which sensitivity is low, for example carbuterol. With the exception of clenhexyl, procaterol, and reproterol all LOQs enable determination of compound concentrations at the MRPL level. CCα and CCβ values are calculated from the within laboratory reproducibility at the level of interest, e.g. 1.0 × VL. As a consequence, CCα and CCβ values are always higher than the validation level at which they are determined. The CCα and CCβ values for the analytes in this study are included in Table 2. In general, for 90% of the analytes the CCα and CCβ values are within a factor of 2 of the VL level at which they were determined. Typical exceptions are carbuterol, clenpenterol, and salmeterol, all belonging to the β-agonist group. Only for salmeterol and, especially, carbuterol is CCα more than a factor of 2 higher than the corresponding VL level.
The robustness of the method was tested by analyzing three identical urine samples fortified at 0.5, 1.0, and 2.0 × VL using the developed procedure with two small deliberate changes in the operating procedure. For the first set of three samples the usual procedure was followed whereas for the second set of samples the SPE column was run dry for 10 min after application of the aqueous extract and before the wash step. For the third set of samples the final residue after evaporation of the solvent was left dry for 30 min before re-dissolving. The results at each VL level were determined and if these were within 3 times the previously determined reproducibility for the original method then the method was regarded as robust. The selectivity of the method is the ability to discriminate between the analyte, the matrix, and any chemical or instrumental noise and was determined by analysis of 20 blank urine samples. The chromatograms obtained from the blank samples were monitored for the analytes of interest. Because no peaks (>LOQ or >3 times the repeatability) were detected in these blank samples except for endogenous compounds, it is concluded that the method is selective. The specificity of the method is the ability to discriminate between the analytes of interest and other related substances that may be isomers, metabolites, or degradation products, or other small molecules that may be present in the matrix. To test the method’s specificity urine samples were fortified with approximately 200 veterinary drugs and pesticides in addition to the target analytes at 0.5, 1.0, and 2.0 × the VL. The chromatograms were monitored for peaks that can potentially interfere with the analytes of interest and the results compared with those obtained from samples fortified with only the target analytes and taking into account the reproducibility of the method. Because the results for urine samples fortified with additional compounds were within 3 times the reproducibility of the method, the method is regarded as specific. Even in the region were cortisol and cortisone elute, determination of other analytes at 0.5× the VL level is not hampered by the often higher cortisol/cortisone concentrations. Finally, stability experiments were carried out for all compounds in extracts stored at −20 °C and for thiazide diuretics in urine at room temperature. Analyses of sample extracts fortified at 0.5, 1.0, and 2.0 times the VL level and stored for four weeks at −20 °C showed that all the analytes were stable during this period. Recovery at the three VL levels ranged from 65 to 147% with average values of 94 ± 22%, 101 ± 18%, and 109 ± 16% at the three VL levels. Methylprednisolone was an exception, because recovery was below 80% at all three VL levels. Because thiazide diuretics are reported to be unstable in urine, because of hydrolysis, an additional stability experiment was performed [43]. Blank urine samples were spiked with thiazide diuretics at the VL level and analysed after storage for 1, 4, and 9 days at room temperature. The results show that althiazide, especially, and bendroflumethiazide and cyclothiazide are not stable at room temperature for prolonged periods. For these compounds, average recoveries after 4 and 9 days at room temperature were 60% and 10%, respectively. For the other thiazides the average recoveries after 4 and 9 days were 80% and 60%, respectively.
Conclusions
A generic sample-preparation procedure was combined with solid-phase extraction tailored for the simultaneous isolation and purification of glucocorticosteroids, β2-agonists, thiazide diuretics, and narcotics and stimulants in urine. All compounds are determined using high-resolution liquid chromatography in combination with time-of-flight mass spectrometry using positive electrospray ionisation. However, for the best sensitivity and quantitative determination the thiazide diuretics are best analysed using negative electrospray ionisation. The results show that with the exception of the β2-agonists clenhexyl, procaterol, and reproterol all compounds can be detected below the respective MRPL values. The results for linearity, repeatability, within-lab reproducibility, and accuracy show that the method is capable of quantitative screening for >90% of the individual compounds. Notable exceptions are clenpenterol, zilpaterol, and salmeterol for which repeatability and reproducibility were >20% and clenpenterol and carbuterol for which the accuracy was just outside the 70–140% acceptability window. If qualitative screening is sufficient instrumental analysis can be limited to positive electrospray ionisation, because all the analytes including the thiazides can be detected at the respective MRPL levels in positive-ion mode. The results show that application of high-resolution liquid chromatography–time-of-flight mass spectrometry in combination with general extraction and purification procedures is suitable for unification of screening procedures for prohibited small-molecule substances. Moreover, the full-scan accurate-mass data sets obtained still enable retrospective examination for emerging doping agents, without re-analysis of the samples. These benefits should allow doping control laboratories to free resources for new classes of banned substances.
References
World Anti-Doping Agency (2009) The world anti-doping code. The 2009 prohibited list. International Standard, Montreal, Canada
Bowers LD (2009) Ann Rev Anal Chem 2:485–507
McDonald M, Granelli K, Sjöberg P (2007) Anal Chim Acta 588:20–25
Mazzarino M, Turi S, Botré F (2008) Anal Bioanal Chem 390:1389–1402
Politi L, Morini L, Polettini A (2007) Clin Chim Acta 386:46–52
Mazzarino M, Botré F (2006) Rapid Commun Mass Spectrom 20:3465–3476
Van der Heeft E, Bolck YJC, Beumer B, Nijrolder AWJM, Stolker AAM, Nielen MWF (2009) J Am Soc Mass Spec 20:451–463
Kellman M, Muenster H, Zomer P, Mol H (2009) J Am Soc Mass Spectrom. doi:10.1016/j.jasms.2009.05.010. Accepted for publication May 22, 2009
Georgakopoulos CG, Vonaparti A, Stamou M, Kiousi P, Lyris E, Angelis YS, Tsoupras G, Wuest B, Nielen MWF, Panderi I, Koupparis M (2007) Rapid Commun Mass Spectrom 21:2439–2446
Stolker AAM, Rutgers P, Oosterink E, Lasaroms JJP, Peters RJB, Van Rhijn JA, Nielen MWF (2007) Anal Bioanal Chem 391:2309–2322
Kaufmann A, Butcher P, Maden K, Widmer M (2007) Anal Chim Acta 586:13–21
Kaufmann A, Butcher P, Maden K, Widmer M (2008) J Chromatogr A 1194:66–79
Mol HGJ, Plaza-Bolanos P, Zomer P, De Rijk ThC, Stolker AAM, Mulder PPJ (2008) Anal Chem 80:9450–9459
Peters RJB, Bolck YJC, Rutgers P, Stolker AAM, Nielen MWF (2009) doi:10.1016/j.chroma.2009.04.027. Accepted for publication in J Chromatogr A
Taylor RL, Grebe SK, Singh RJ (2004) Clin Chem 50:2345–2352
Catlin DH, Sekera MH, Arens BD, Starcevic B, Chang YC, Hatton CK (2004) Rapid Commun Mass Spectrom 18:1245–1249
Argenti D, Jensen BK, Hensel R, Bordeaux K, Schleimer R, Bickel C, Heald D (2001) J Clin Pharmacol 51:400–409
Pearce RE, Leeder JS, Kearns GL (2006) Drug Metab Dispos 34:1035–1040
Piram A, Salvador A, Gauvrit JY, Lanteri P, Faure R (2007) Talanta 74:1463–1475
Reddy IM, Beotra A, Jain S, Ahi S (2009) Indian J Pharmacol 41:80–86
Touber ME, Van Engelen MC, Georgakopoulus C, Van Rhijn JA, Nielen MWF (2007) Anal Chim Acta 586:137–146
Ojanperä S, Pelander A, Pelzing M, Kreb I, Cuori E, Ojanperä I (2006) Rapid Commun Mass Spectrom 20:1161–1167
Kolmonen M, Leinonen A, Pelander A, Ojanperä I (2007) Anal Chim Acta 585:94–102
World Anti-Doping Agency (2003) Technical Document TD2003IDCR
Fitch KD (2006) Clin. Rev Allergy Immunol 31:259–268
Kindermann W (2007) Sports Med 37:95–102
Kang MJ, Hwang YH, Kim DH (2007) Rapid Commun Mass Spectrom 21:252–264
Virus ED, Sobolevsky TG, Rodchenkov GD (2008) J Mass Spectrom 43:949–957
Beyer J, Bierl A, Peters FT, Maurer HH (2005) Ther. Drug Monit 27:509–520
Morra V, Davit P, Capra P, Vincenti M, Di Stilo A, Botré F (2006) J Chromatogr A 1135:219–229
Deventer K, Delbeke FT, Roels K, Van Eenoo P (2002) Biomed Chromatogr 16:529–535
Politi L, Morini L, Polettini A (2007) Clin Chim Acta 386:46–52
Ventura R, Roig M, Montfort N, Sáez P, Bergés R, Segura J (2008) Eur J Mass Spectrom 14:191–200
Strano-Rossi S, Colamonici C, Botré F (2008) Anal Chim Acta 606: ■■217–222■■
Strano-Rossi S, Molaioni F, Botré F (2005) J Anal Toxicol 29: ■■319–326■■
Pragst F (2007) Anal Bioanal Chem 388:1393–1414
Thörngren JO, Ostervall F, Garle M (2008) J Mass Spectrom 43:980–992
World Anti-Doping Agency (2009) International Standard for Laboratories. Montreal, Canada
Official Journal of the European Communities (2002) L221 of 17 August 2002, Commission Decision 2002/657/EC of 12 August 2002 concerning the performance of analytical methods and the interpretation of results. Brussels, Belgium
Laures AMF, Wolff JC, Eckers Ch, Borman PhJ, Chatfield MJ (2007) Rapid Commun Mass Spectrom 21:529–535
Bristow T, Constantine J, Harrison M, Cavoit F (2008) Rapid Commun Mass Spectrom 22:1213–1222
World Anti-Doping Agency (2009) WADA Technical Document –TD2009DL
Goebel C, Trout GJ, Kazlauskas R (2004) Anal Chim Acta 502:65–74
Acknowledgement
This study was supported financially by the World Anti-Doping Agency and co-financed by the Dutch Ministry of Agriculture, Nature, and Food Quality (project # 87163901).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Peters, R.J.B., Oosterink, J.E., Stolker, A.A.M. et al. Generic sample preparation combined with high-resolution liquid chromatography–time-of-flight mass spectrometry for unification of urine screening in doping-control laboratories. Anal Bioanal Chem 396, 2583–2598 (2010). https://doi.org/10.1007/s00216-010-3484-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-010-3484-3