
Abstract
A high-throughput Förster resonance energy transfer (FRET) study was performed on the approximately 100 amino acids long N-terminal domain of the photosynthetic complex CP29 of higher plants. For this purpose, CP29 was singly mutated along its N-terminal domain, replacing one-by-one native amino acids by a cysteine, which was labeled with a BODIPY fluorescent probe, and reconstituted with the natural pigments of CP9, chlorophylls and xanthophylls. Picosecond fluorescence experiments revealed rapid energy transfer (~20–70 ps) from BODIPY at amino-acid positions 4, 22, 33, 40, 56, 65, 74, 90, and 97 to Chl a molecules in the hydrophobic part of the protein. From the energy transfer times, distances were estimated between label and chlorophyll molecules, using the Förster equation. When the label was attached to amino acids 4, 56, and 97, it was found to be located very close to the protein core (~15 Å), whereas labels at positions 15, 22, 33, 40, 65, 74, and 90 were found at somewhat larger distances. It is concluded that the entire N-terminal domain is in close contact with the hydrophobic core and that there is no loop sticking out into the stroma. Most of the results support a recently proposed topological model for the N-terminus of CP29, which was based on electron-spin-resonance measurements on spin-labeled CP29 with and without its natural pigment content. The present results lead to a slight refinement of that model.
Similar content being viewed by others
Introduction
Photosynthesis in higher plants is performed by two major complexes, photosystem (PS) I and II, which are composed of a core complex and an antenna part. The PSII antenna (for an overview of the PSII antenna; see, e.g., van Amerongen and Croce 2007; van Amerongen and Dekker 2003; van Amerongen and van Grondelle 2001) consists of many light-harvesting pigment–protein complexes that are organized around the reaction center, and it comprises the major light-harvesting complex II (LHCII) which exists as trimers, and the minor antenna proteins CP24, CP26 and CP29. All these complexes bind Chls a and b and various xanthophylls. After excitation of these pigments by light, excitation energy is transferred through the antenna on a picosecond time scale, eventually leading to charge separation in the reaction center. Although light-harvesting is very efficient under low-light conditions, tight regulation and protection mechanisms (for instance, nonphotochemical quenching or NPQ) are necessary to protect PSII against destructive free radicals, being produced in excess light. The PSII antenna is also actively involved in this process, and it is has been proposed that a conformational change of LHCII is to a large extent responsible for NPQ (Pascal et al. 2005; Ruban et al. 2007), although it has also been argued that a similar conformational change in the minor antenna complexes, like, e.g., CP29, might lead to NPQ (Mozzo et al. 2008). On the other hand, it has been proposed that the minor antenna complexes participate in NPQ via radical cation formation on zeaxanthin molecules (in particular in CP29; Ahn et al. 2008; Avenson et al. 2008; Cheng et al. 2008; Wormit and Dreuw 2007).
The structures of most of the pigment–protein complexes constituting PSII have been determined with the exception of those of minor antenna proteins. The structures of their transmembrane parts are thought to be very similar to those of LHCII because of high sequence homology (see, e.g., van Amerongen and Croce 2007) and spectroscopic similarities (Cinque et al. 2000a, b; Croce et al. 2003a, b; Gradinaru et al. 1998, 2000; Mozzo et al. 2008; Pascal et al. 1999; Salverda et al. 2003; Zucchelli et al. 1994). But CP29 has, for instance, a very long N-terminal domain of around 100 amino acids, much longer than the N-terminal domain of LHCII, and obviously its structure cannot be completely modeled based on analogy with the structure of LHCII. Up to now, the exact function of this N-terminal stretch is unknwon, although it has been found that under cold stress the N-terminus becomes phosphorylated at the threonine residue in position 81 (Testi et al. 1996), after which the protein undergoes a conformational change (Croce et al. 1996; Nilsson et al. 1997).
Among the three minor light-harvesting proteins, CP29 is the best characterized one. As a product of the Lhcb4 gene, CP29 is the largest of the outer antenna complexes, but also the one binding the lowest number of pigments: 6 Chls a, 2 Chls b, and 2.3 xanthophyll molecules, located in three binding sites (Caffarri et al. 2007). The major differences from LHCII are the presence of violaxanthin in the L2 binding site and the low concentration of Chls b in the A-C helix domain, which makes CP29 the complex with the highest Chl a/b ratio amongst the Lhcb antennae. This is probably related to the fact that CP29 in the structure of the PSII supercomplex is located between the LHCII trimers and the core (Dekker and Boekema 2005), and that it represents an obligatory “intermediate station” for the transfer of the excitation energy from the periphery of the PSII supercomplex to the PSII core.
Recently, our group has started a research program to study the structure and dynamics of CP29, starting with the N-terminal domain, by labeling and reconstituting this pigment–protein complex. A single cysteine replaces individual amino acids along the N-terminal region, which is labeled with either a fluorescence (van Oort et al. 2009) or a spin-label probe (Kavalenka et al. 2009).
Labeling CP29 at amino acid positions 4, 40 and 97 with a fluorescent TAMRA probe allowed the measurement of ultrafast excitation energy transfer (FRET) from the probe to the Chl (van Oort et al. 2009). It turned out that transfer from the probe attached at position 4 was as fast (~20 ps) as from the probe attached at position 97, which is located close to the transmembrane pigment-containing part of the protein, demonstrating that the last stretch of the N-terminus is folded back on the hydrophobic core. Position 40 appeared to be farther away, but some uncertainty remained about the exact transfer time. A complicating factor turned out to be the fact that the fluorescence kinetics were nonexponential, and even for the label at position 97, which is supposed to be very close to the Chls, a component of several nanoseconds was observed, much longer than one might expect for correctly folded CP29.
In a separate ESR study, we labeled the protein at 10 different positions with an MTS-SL spin label and measured and simulated all ESR spectra both in the absence and presence of the chlorophyll and xanthophyll pigments. This led to a schematic structural model in which position 4 is indeed close to the hydrophobic core, whereas a flexible loop, containing positions 65, 82 and 90, is proposed to extend out of the protein. The rest of the N-terminal domain seemed to be rigid and close to the hydrophobic part of the protein with the exception of a region around position 15, which also appeared to be flexible and farther away from the hydrophobic domain.
In the present study, we have extended our time-resolved fluorescence approach by attaching a fluorescent probe at nearly all the positions that were also spin-labeled before, in order to test the proposed topological organization of the N-terminus and to obtain distance constraints for the various parts of the N-terminus with respect to the hydrophobic core. Instead of the TAMRA label that was used before, a smaller, more inert BODIPY-fluorescent label (Loudet and Burgess 2007) was applied this time, to decrease the probability that the label is influencing the protein structure.
Materials and methods
Mutagenesis, labeling and pigment reconstitution
Various single cysteine mutants of CP29 were created by replacing the naturally occurring amino acids at position 4 (glycine), 15 (serine), 22 (threonine), 33 (serine), 40 (serine), 56 (alanine), 65 (serine), 74 (leucine), 90 (serine), or 97 (serine) by cysteine, while in all cases the naturally occurring cysteine at position 108 was replaced by an alanine, using methods as described before (Spruijt et al. 2000; van Oort et al. 2009). The unlabeled protein (C108A) served as a wild-type-like reference (WT). CP29 apoproteins (proteins without pigments) containing a single cysteine moiety were labeled with MTS-BODIPY (4,4-Difluro-8-methylmethanethiosulfonyl-1,3,5,7-tetramethyl-4-bora-3a,4a-diaza-5-indacene; TRC, Toronto, Canada) (Nagai and Thøgersen 1987; Paulsen et al. 1990; Spruijt et al. 2000). Removal of non-site-specifically bound label and subsequent pigment reconstitution was done as described in (Croce et al. 2003a, b; Spruijt et al. 2000). The labeling-efficiency and protein concentrations were determined by UV-VIS absorption. The average labeling efficiency was 75 ± 32% (number of labels per number of proteins). The amount of non-specifically bound label was less than 1% for WT without Cys. All CP29 complexes are monomeric as assessed by sucrose gradient centrifugation.
Steady-state spectra
Steady-state absorption spectra were recorded on a Varian Cary 5E spectrophotometer. Steady-state fluorescence emission spectra (475 or 515 nm excitation) were recorded on a Spex-Fluorolog 3.2.2 spectrofluorimeter (Jobin-Yvon).
Time-correlated single-photon counting
Time-correlated single-photon counting (TCSPC) measurements were performed using a homebuilt setup as described in Borst et al. (2005) and Visser et al. (2008), using the same methodology as in van Oort et al. (2009). Excitation pulse energies of a few pJ were used, the excitation wavelength was set to 475 nm, while the pulse duration was less than 0.2 ps with a repetition rate of 3.8 MHz. Fluorescence was detected by a microchannel plate photomultiplier, and arrival times were stored in 4,096 channels of a multichannel analyzer with a channel width of 5 ps. The excitation density was adjusted to obtain a photon count rate of 3 × 104 per second and care was taken to minimize data distortion (Vos et al. 1987). For each sample, data were acquired during 10 repeated sequences of gathering 10 s parallel and 10 s perpendicularly polarized fluorescence photons. The instrument response function (FWHM ~35 ps) was obtained with erythrosine B in water, with a known fluorescence lifetime of 80 ps (Lin et al. 1999). Data were corrected for background signal by subtracting the signal of DM buffer measured directly after each CP29 sample. Fluorescence decay curves were fitted to a sum of exponential decay functions and convoluted with the instrument response function. The quality of the fit was judged by the chi-square value and by the quality of the residuals and autocorrelation of these residuals.
Streak-camera measurements and data analysis
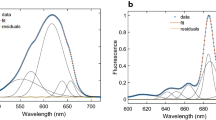
Two-dimensional images of fluorescence intensity as a function of time and wavelength were recorded with a streak-camera setup with a spectral resolution of typically 6 nm, and a temporal resolution of 4 ps (van Oort et al. 2009; van Stokkum et al. 2008). The time window for the experiments was either 800 or 160 ps, and the spectral window was 260 nm with the central wavelength located at 650 nm. Excitation pulses of 515 nm were applied with a repetition frequency of 253 kHz and a pulse width of 130 fs. A 530-nm cut-off filter was used to avoid the recording of a scatter peak. The data were corrected for jitter, averaged, and background- and shading-corrected using the High Performance Digital Temporal Analyzer (HPDTA 8.3.0; Hamamatsu Photonics). The streak-camera images were sliced up into time traces of 2.6 nm width and these traces were fitted to a sum of exponentials using global analysis with software described in Mullen and van Stokkum (2007). The resulting wavelength-dependent amplitudes provide the decay-associated spectra (DAS) shown in the “Results”.
Distance calculations based on Förster resonance energy transfer
The obtained excitation energy transfer rates were used to calculate distances between donor and acceptor molecules, using the Förster resonance energy transfer theory (Förster 1949). The rate of energy transfer from a donor to an acceptor k T(R) is given by the Förster equation (Lakowicz 2006)
where τD is the fluorescence lifetime of the donor in the absence of acceptor (4.9 ns), R is the donor to acceptor distance, and R 0 is the Förster radius, which is given by
where n is usually considered to be the refractive index of the medium between donor and acceptor and a value of 1.4 is often taken (Lakowicz 2006) (for a detailed discussion, see, Knox and van Amerongen 2002), Q D is the fluorescence quantum yield of the donor (0.57), experimentally determined by using Rhodamine 6G in ethanol (Exciton, Dayton, Ohio) with fluorescence quantum yield 0.95 as a reference (Fischer and Georges 1996), and κ2 the orientation factor, depending on the relative orientation of donor and acceptor. κ2 = 2/3 is a generally accepted value for random relative orientation of donor and acceptor molecules (Lakowicz 2006) and this value is also used here. J(λ), the overlap integral between the fluorescence spectrum of the donor and the absorption spectrum of the acceptor, is defined as
The overlap integral is calculated from the emission spectrum of BODIPY labeled apoprotein and the absorption spectrum of Chl a (Croce et al. 2003a). This leads to a value of R 0 = 4.3 nm.
Results
Steady-state spectra
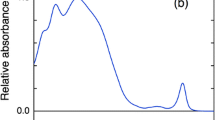
All labeled CP29 holoproteins (proteins reconstituted with pigments) show absorption spectra that are identical to each other in the Qy region, demonstrating that the proteins have folded correctly (see, e.g., Bassi et al. 1999), whereas they show a significant absorption increase around 514 nm as compared to unlabeled C108A reconstituted protein, due to BODIPY absorption (see Fig. 1 for a typical example). Upon 515-nm excitation, the steady-state emission spectra of labeled, mutated CP29 holoproteins all show an increase in Chl fluorescence when compared to unlabeled CP29 after normalization to the Chl concentration, due to FRET from BODIPY to Chl. Also, some BODIPY fluorescence can be observed around 520 nm for all labeled holoproteins, demonstrating that not all excitations are transferred to the Chl molecules.

Normalized absorption spectra of unlabeled WT (red), CP29 holoprotein (black), and CP29 apoprotein (green), both labeled at position 90. Also, the fluorescence spectrum (blue) of the latter, exciting at 475, is presented with normalized intensity
TCSPC
For measuring the fluorescence decay of BODIPY attached to the apoprotein (at 520 nm), the excitation wavelength was set to 475 nm. All apoprotein-specimens gave rise to BODIPY fluorescence decay curves that could be fitted to a sum of three exponentials with decay times of 0.47, 3.1, and 6.5 ns. The amplitudes are similar for all components in all cases, i.e., close to 1/3 (data not shown). The fluorescence decay of BODIPY attached to the holoprotein was much faster in all cases, and a typical example is given in Fig. 2. To fit the fluorescence decay curves of the CP29 holoprotein mutants, an additional short lifetime component was needed in all cases. Linking the decay times of the three slowest components for all samples in a global analysis procedure, while leaving the fastest component free, resulted in good fits in all cases (Fig. 2). The slowest decay times were 0.36, 2.3, and 6.3 ns. Depending on the position along the N-terminal domain, the BODIPY fluorescence decay time of the fast component varied from around 20 to 70 ps (Table 1).

Normalized fluorescence decay curves of CP29 apoprotein (magenta) and holoprotein (green), labeled at position 15, including the multi-exponential fits (black) (see Table 1)
Like before (van Oort et al. 2009), the shortest decay time is ascribed to excitation energy transfer from BODIPY to the Chl molecules and the fastest times are observed for positions 4C, 56C, and 97C. In all cases, the amplitude of the fast component is at least 64%, with an average value of 76%. An extra purification step revealed the presence of a substantial fraction of labeled proteins that were not binding any pigments, and therefore it is concluded that the long lifetime components should mainly be ascribed to labeled apoproteins without pigments. The presence of the long lifetime components in mutant 97C also confirms this conclusion, since this amino acid is supposed to be very close to the Chls in the holoprotein and no slow excitation energy transfer is expected. Possibly, in some cases, the fitted lifetime of 363 ps also reflects some energy transfer. For instance, for the S15A mutant, the value of α2 is significantly higher than the values for α3 and α4, unlike what is observed for all apoproteins and most holoproteins. Nevertheless, most of the excitation energy transfer occurs on a much faster time scale and can be described with a 55 ps transfer time in the case of S15A holoprotein.
Streak-camera measurements
Time-resolved measurements were also performed in some cases with the streak-camera setup. This allows the simultaneous recording of donor (BODIPY) fluorescence decay and acceptor (Chl) fluorescence rise with high time resolution. Two-dimensional fluorescence images (intensity as a function of time and wavelength) were obtained using 515-nm excitation, 520- to 780-nm detection and either the 160- or the 800-ps time window. All decay traces were fitted globally to a sum of exponentials. Figure 3 shows the decay associated spectra (DAS) of the fluorescence kinetics of mutant 74 as a typical example. In the left panel of Fig. 3, the fastest component is fixed to 62 ps, which is the value that was found for the TCSPC experiments. It clearly shows the characteristics of excitation energy transfer, i.e., a positive spectrum in the donor region and a negative one in the acceptor region. In the right panel, a four-exponential fit shows that the 62-ps transfer component is most likely due to several transfer components, and instead of a 62-ps transfer DAS, two spectra are obtained with corresponding times of 20 and 122 ps. The 122-ps DAS shows the clear characteristics of energy transfer (which would correspond to a distance of 2.4 nm, using the same methods as applied to obtain the distances in Table 1), but for the 20 ps this is less obvious.

DAS resulting from global analysis of the streak image of the holoprotein labeled at position 74, using an 800-ps time window. Three-component analysis (left panel) transfer component fixed at 62 ps (black), and decay components of 170 ps (red) and 3.2 ns (green). Four-component analysis (right panel) transfer components (black) of 20 ps (dashed) and 122 ps (solid), and decay components of 172 (red) and 3.2 ns (green). Note that in both cases the red spectrum has been fixed to 0 below 600 nm
Note that the transfer times are much longer than the transfer times from Chls b and xanthophylls to Chls a which are typically below 1 ps (Croce et al. 2003a, b; Gradinaru et al. 1998, 2000) and therefore the rise times are entirely due to transfer from the label to nearby pigments and not to energy transfer between pigments. The second and third spectrum decay with time constants of 170/172 ps and 3.2 ns, respectively, without showing any signs of energy transfer from the BODIPY to the Chl region. These slower times are less accurately determined than in the TCSPC experiments, mainly because of the narrow time window and the lower signal-to-noise ratio.
Discussion
It was already observed in our previous study of CP29 labeled with TAMRA (van Oort et al. 2009) that, besides the fast decay time that clearly corresponds to excitation energy transfer, slower decay components were also present, ranging from a few hundred ps to several ns. It has been discussed to which extent this variation in lifetimes might have been due to variability in the structure of the N-terminal domain. Also in the present study, a large variation of lifetimes is observed. However, it is now argued that the measured samples contain a fraction of labeled CP29 apoproteins without any pigments bound, which are to a large extent responsible for the long lifetime components (several hundreds of ps and longer). In all cases, the donor fluorescence decay is dominated by a component of several tens of ps, which is absent for all apoproteins and which can be entirely ascribed to excitation energy transfer from BODIPY to Chl molecules.
In this study, we have efficiently labeled the N-terminal domain of CP29 at ten different positions, using the relatively small fluorescence probe MTS-BODIPY. This probe is smaller than the TAMRA probe that was used before (van Oort et al. 2009), and moreover it contains less charges. Therefore, it is less likely that the BODIPY probe leads to any distortion of the CP29 structure. It has been shown before that, when TAMRA is attached at positions 4 or 97, very fast resonance energy transfer occurs to the Chl molecules, characterized by nearly identical transfer times (19–20 ps). From these results, it was concluded that the N-terminus is folded back on the hydrophobic core since position 97 is known to be located directly next to the part where the Chls are bound. In order to explain a similarly fast transfer time from position 4, this position must also be very close to the hydrophobic core, containing the pigments. Similar results have now been obtained for BODIPY: the lifetimes are 27 ps for position 4 and 32 ps for position 97. Note that the lifetimes obtained with the different labels cannot be directly compared to each other because the Förster radius differs. When the TAMRA was attached to position 40, the transfer was clearly slower than for the other two positions. The same trend is now observed for BODIPY attached to position 40, which shows a transfer time of 61 ps.
In order to estimate the distance between the label and the most nearby Chl molecule for the various mutants, use is made of the Förster equation (see “Materials and methods”). The lifetime of 27 ps for position 4 corresponds to a distance of 1.69 nm, slightly shorter than the distance of 1.73 nm obtained for position 97. These distances should be treated with some caution for several reasons. First of all, not all parameters in the Förster equation are precisely known; secondly, it is not known how the cysteine is exactly positioned with respect to the label; and thirdly, it is not known to which Chl(s) the label is transferring its excitation energy. To get a feeling about the accuracy of the calculations, the distance was determined from the crystal structure of LHCII, using the Cα-atom of the corresponding amino acid in LHCII (alanine 56) and the center of the most nearby Chl (Chl a4, using the nomenclature of Kühlbrandt et al. 1994). The obtained value is 1.36 nm. This is smaller than the value of 1.73 nm, estimated from the transfer time. However, the difference between these values is well within the distance from the center of the BODIPY label to the Cα-atom of the cysteine (that replaces the alanine), which is approximately 0.7 nm. It should be realized that the presence of more than one acceptor molecule does not substantially influence the calculated distance between donor and acceptor molecule. If the transfer to the other Chls is also taken into account for position 97, the calculated apparent distance would be 1.31 nm instead of 1.36 nm. Therefore, the estimated distance of 1.7 nm, both for positions 4 and 97, reflect the fact that the amino acids at these positions are located very close to the hydrophobic core of CP29 where the Chl molecules are located.
The distance between the label and the nearest Chl was estimated for all measured CP29 mutants and the results are given in Table 1. The striking result is that for all label positions the value of R falls within the narrow interval from 1.7 to 2.1 nm. This makes clear that the entire N-terminal domain is folded back on the hydrophobic core. For positions 4, 33, 40, 56, and 97, it was already concluded from the previous ESR measurements that the corresponding amino acids are probably close to the hydrophobic core, because the ESR spectrum of an electron-spin label attached to each of these positions is significantly different in the presence and absence of pigments, and moreover the label appears to be rather immobilized. But when the label is attached to a cysteine at positions 15, 65, and 90, it appears to be more flexible and the spectrum is less dependent on the presence/absence of pigments. Based on these results, it was proposed in a cartoon-like model that two loops were sticking out into the stroma, a short one containing amino acid 15 and a longer one containing amino acids 65 and 90. We have to conclude now that these relatively flexible loops are also not sticking out but are lying flat on the hydrophobic core most of the time.
Abbreviations
- Chl:
-
Chlorophyll
- CP:
-
Chlorophyll protein
- DAS:
-
Decay associated spectrum/spectra
- DM:
-
Dodecyl β-D-maltoside
- ESR:
-
Electron spin resonance
- FWHM:
-
Full width at half maximum
- FRET:
-
Förster resonance energy transfer
- MTS:
-
Methanethiosulfonate
- NPQ:
-
Non-photochemical quenching
- PS:
-
Photosystem
- R0 :
-
Förster distance
- TCSPC:
-
Time correlated single photon counting
- WT:
-
Wild-type
References
Ahn TK, Avenson TJ, Ballottari M, Cheng Y-C, Niyogi KK, Bassi R, Fleming GR (2008) Architecture of a charge-transfer state regulating light harvesting in a plant antenna protein. Science 320:794–797
Avenson TJ, Ahn TK, Zigmantas D, Niyogi KK, Li Z, Ballottari M, Bassi R, Fleming GR (2008) Zeaxanthin radical cation formation in minor light-harvesting complexes of higher plant antenna. J Biol Chem 283:3550–3558
Bassi R, Croce R, Cugini D, Sandona D (1999) Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc Natl Acad Sci USA 96:10056–10061
Borst JW, Hink MA, van Hoek A, Visser AJWG (2005) Effects of refractive index and viscosity on fluorescence and anisotropy decays of enhanced cyan and yellow fluorescent proteins. J Fluoresc 15:153–160
Caffarri S, Passarini F, Bassi R, Croce R (2007) A specific binding site for neoxanthin in the monomeric antenna proteins CP26 and CP29 of Photosystem II. FEBS Lett 581:4704–4710
Cheng YC, Ahn TK, Avenson TJ, Zigmantas D, Niyogi KK, Ballottari M, Bassi R, Fleming GR (2008) Kinetic modeling of charge-transfer quenching in the CP29 minor complex. J Phys Chem B 112:13418–13423
Cinque G, Croce R, Bassi R (2000a) Absorption spectra of chlorophyll a and b in Lhcb protein environment. Photosynth Res 64:233
Cinque G, Croce R, Holzwarth A, Bassi R (2000b) Energy transfer among CP29 chlorophylls: calculated Forster rates and experimental transient absorption at room temperature. Biophys J 79:1706–1717
Croce R, Breton J, Bassi R (1996) Conformational changes induced by phosphorylation in the CP29 subunit of photosystem II. Biochemistry 35:11142–11148
Croce R, Müller MG, Bassi R, Holzwarth AR (2003a) Chlorophyll b to Chlorophyll a energy transfer kinetics in the CP29 antenna complex: a comparative femtosecond absorption study between native and reconstituted proteins. Biophys J 84:2508–2516
Croce R, Müller MG, Caffarri S, Bassi R, Holzwarth AR (2003b) Energy transfer pathways in the minor antenna complex CP29 of photosystem II: a femtosecond study of carotenoid to chlorophyll transfer on mutant and WT complexes. Biophys J 84:2517–2532
Dekker JP, Boekema EJ (2005) Supramolecular organization of thylakoid membrane proteins in green plants. Biochim Biophys Acta 1706:12–39
Fischer M, Georges J (1996) Fluorescence quantum yield of rhodamine 6G in ethanol as a function of concentration using thermal lens spectrometry. Chem Phys Lett 260:115–118
Förster T (1949) Experimentelle Und Theoretische Untersuchung Des Zwischenmolekularen Ubergangs Von Elektronenanregungsenergie. Z Naturforsch 4a:321–327
Gradinaru CC, Pascal AA, van Mourik F, Robert B, Horton P, van Grondelle R, van Amerongen H (1998) Ultrafast evolution of the excited states in the chlorophyll a/b complex CP29 from green plants studied by energy-selective pump-probe spectroscopy. Biochemistry 37:1143–1149
Gradinaru CC, van Stokkum IHM, Pascal AA, van Grondelle R, van Amerongen H (2000) Identifying the pathways of energy transfer between carotenoids and chlorophylls in LHCII and CP29. A multicolor, femtosecond pump-probe study. J Phys Chem B 104:9330–9342
Kavalenka AA, Spruijt RB, Wolfs CJAM, Strancar J, Croce R, Hemminga MA, van Amerongen H (2009) Site-directed spin-labeling study of the light-harvesting complex CP29. Biophys J 96:3620–3628
Knox RS, van Amerongen H (2002) Refractive index dependence of the Förster resonance excitation transfer rate. J Phys Chem B 106:5289–5293
Kühlbrandt W, Wang DN, Fujiyoshi Y (1994) Atomic model of plant light-harvesting complex by electron crystallography. Nature 367:614–621
Lakowicz JR (2006) Principles of fluorescence spectroscopy, 3rd edn. Springer, New York
Lin HJ, Szmacinski H, Lakowicz JR (1999) Lifetime-based pH sensors: indicators for acidic environments. Anal Biochem 269:162–167
Loudet A, Burgess K (2007) BODIPY dyes and their derivatives: syntheses and spectroscopic properties. Chem Rev 107:4891–4932
Mozzo M, Passarini F, Bassi R, van Amerongen H, Croce R (2008) Photoprotection in higher plants: the putative quenching site is conserved in all outer light-harvesting complexes of Photosystem II. Biochim Biophys Acta 1777:1263–1267
Mullen KM, van Stokkum IHM (2007) TIMP: an R package for modeling multi-way spectroscopic measurements. J Stat Softw 18:46
Nagai K, Thøgersen HC (1987) Synthesis and sequence-specific proteolysis of hybrid proteins produced in Escherichia coli. Meth Enzymol 153:461–481
Nilsson A, Stys D, Drakenberg T, Spangfort MD, Forsen S, Allen JF (1997) Phosphorylation controls the three-dimensional structure of plant light harvesting complex II. J Biol Chem 272:18350–18357
Pascal A, Gradinaru C, Wacker U, Peterman E, Calkoen F, Irrgang K-D, Horton P, Renger G, van Grondelle R, Robert B, van Amerongen H (1999) Spectroscopic characterization of the spinach Lhcb4 protein (CP29), a minor light-harvesting complex of photosystem II. Eur J Biochem 262:817–823
Pascal AA, Liu ZF, Broess K, van Oort B, van Amerongen H, Wang C, Horton P, Robert B, Chang WR, Ruban A (2005) Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436:134–137
Paulsen H, Rümler U, Rüdiger W (1990) Reconstitution of pigment-containing complexes from light-harvesting chlorophyll a/b-binding protein overexpressed in Escherichia coli. Planta 181:204–211
Ruban AV, Berera R, Ilioaia C, van Stokkum IHM, Kennis JTM, Pascal AA, van Amerongen H, Robert B, Horton P, van Grondelle R (2007) Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 450:575–579
Salverda JM, Vengris M, Krueger BP, Scholes GD, Czarnoleski AR, Novoderezhkin VI, van Amerongen H, van Grondelle R (2003) Energy transfer in light-harvesting complexes LHCII and CP29 of spinach studied with three pulse echo peak shift and transient grating. Biophys J 84:450–465
Spruijt RB, Meijer AB, Wolfs CJAM, Hemminga MA (2000) Localization and rearrangement modulation of the N-terminal arm of the membrane-bound major coat protein of bacteriophage M13. Biochim Biophys Acta 1509:311–323
Testi MG, Croce R, Polverino-De Laureto P, Bassi R (1996) A CK2 site is reversibly phosphorylated in the photosystem II subunit CP29. FEBS Lett 399:245–250
van Amerongen H, Croce R (2007) Structure and function of photosystem II light-harvesting proteins (Lhcb) of higher plants. In: Renger G (ed) Primary processes of photosynthesis, part 1: principles and apparatus, vol 1. Royal Society of Chemistry, Cambridge, pp 329–368
van Amerongen H, Dekker JP (2003) Light-harvesting in photosystem II. In: Green BR, Parson WW (eds) Light-harvesting antennas in photosynthesis, vol 13. Kluwer, Dordrecht, pp 219–251
van Amerongen H, van Grondelle R (2001) Understanding the energy transfer function of LHCII, the major light-harvesting complex of plants. J Phys Chem B 105:604–617
van Oort B, Murali S, Wientjes E, Koehorst RBM, Spruijt RB, van Hoek A, Croce R, van Amerongen H (2009) Ultrafast resonance energy transfer from a site-specifically attached fluorescent chromophore reveals the folding of the N-terminal domain of CP29. Chem Phys 357:113–119
van Stokkum IHM, van Oort B, van Mourik F, Gobets B, van Amerongen H (2008) (Sub)-picosecond spectral evolution of fluorescence studied with a synchroscan streak-camera system and target analysis. In: Aartsma TJ, Matysik J (eds) Biophysical techniques in photosynthesis II, vol 26. Springer, Dordrecht, pp 223–240
Visser NV, Westphal WG, van Hoek A, van Mierlo CPM, Visser AJWG, van Amerongen H (2008) Tryptophan-tryptophan energy migration as a new tool to follow protein folding. Biophys J 95:2462–2469
Vos K, van Hoek A, Visser AJWG (1987) Application of a references convolution method to tryptophan fluorescene in proteins—a refined description of rotational-dynamics. Eur J Biochem 165:55–63
Wormit M, Dreuw A (2007) Quantum chemical insights in energy dissipation and carotenoid radical cation formation in light harvesting complexes. Phys Chem Chem Phys 9:2917–2931
Zucchelli G, Dainese P, Jennings RC, Breton J, Garlaschi FM, Bassi R (1994) Gaussian decomposition of absorption and linear dichroism spectra of outer antenna complexes of photosystem II. Biochemistry 33:8982–8990
Acknowledgments
This work is partially supported by the ‘Stichting voor Fundamenteel Onderzoek der Materie (FOM)’, which is financially supported by the ‘Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO)’. B.v.O. was financially supported by FOM. S.P.L. was supported by the sandwich Programme of Wageningen University and by the European Community (Marie Curie Research Training Network MRTN-CT-2005-019481 “From FLIM to FLIN”). The authors wish to thank Prof. R. Croce for help with the biochemistry and for useful discussions, and Francesca Passarini for help with protein purification.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
The more you see: Spectroscopy in molecular biophysics.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Berghuis, B.A., Spruijt, R.B., Koehorst, R.B.M. et al. Exploring the structure of the N-terminal domain of CP29 with ultrafast fluorescence spectroscopy. Eur Biophys J 39, 631–638 (2010). https://doi.org/10.1007/s00249-009-0519-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00249-009-0519-9